Die Rolle des Adipokins Chemerin und seines Rezeptors CMKLR1 im Hepatozellulären Karzinom
171
0
0
Volltext
(2)
(3)
(4)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
Abbildung
+7
ÄHNLICHE DOKUMENTE
Hence, the slowly sinking and presumably finer particles we measured showing characteristic -enhanced REE concentrations and strong positive Ce anomalies, closely
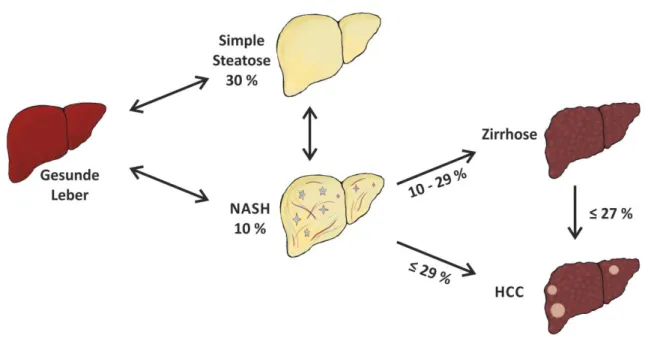
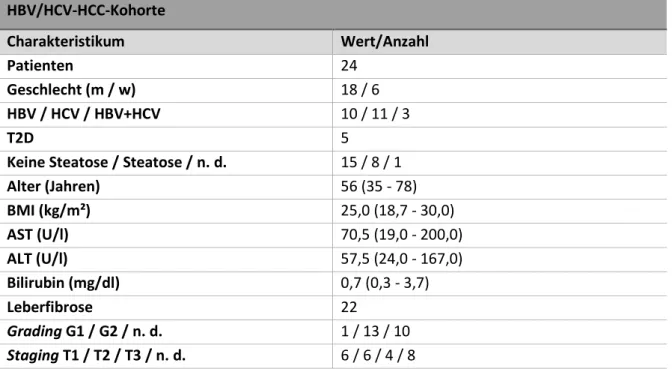
33 34 Fitting to this, also hepatic expression of collagen α I(1) (Coll-I), the most abundant extracellular matrix (ECM) protein of fi brotic liver tissue, and TGF-ß1, a strong pro
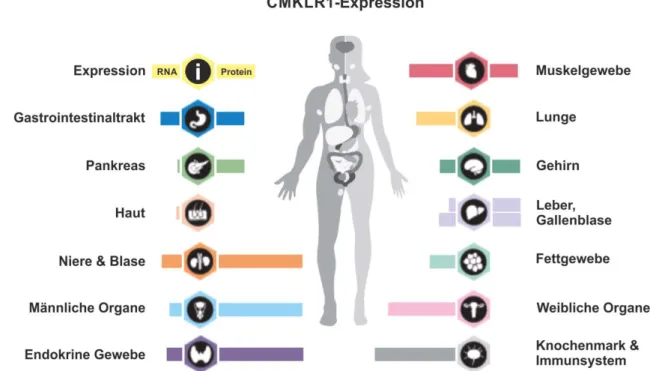
Albeit this shows that chemerin is released from the liver at least in these patients this does not prove that higher hepatic chemerin synthesis translates to increased serum
[r]
European Journal of Taxonomy 374: 1–23 2017 area, rectangular pseudoloculi replaced by 1–2 small, rounded ones whereas near the apices, central transapically elongated
is illustrated and discussed based on populations collected from the Vouga, Mondego and Lis river basins in central Portugal and compared with the type material of Fragilaria
means for developing a first current pulse when all of said stored energy has been transferred out through said secondary winding, said first current pulse being applied to
In accordance one aspect of the invention, the mag- netic disk of the invention comprises plural circumfer- entially spaced data sectors that include at least two sets
