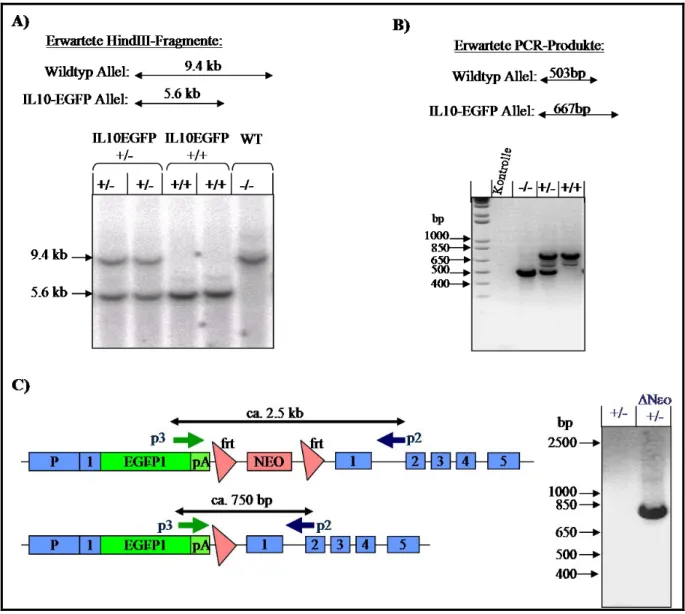
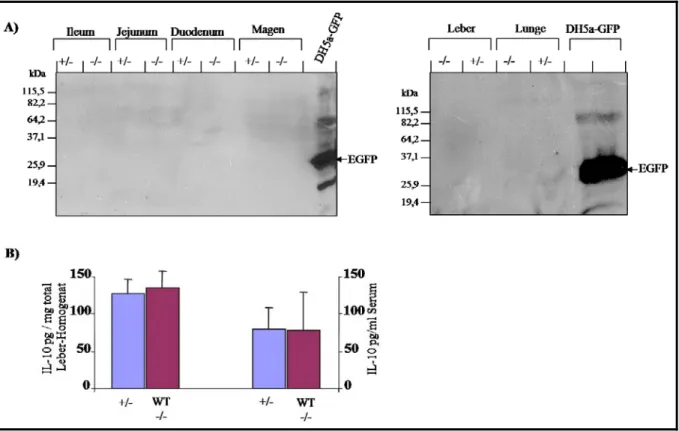
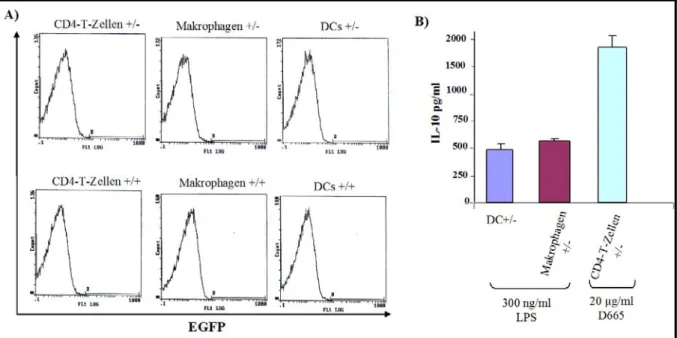
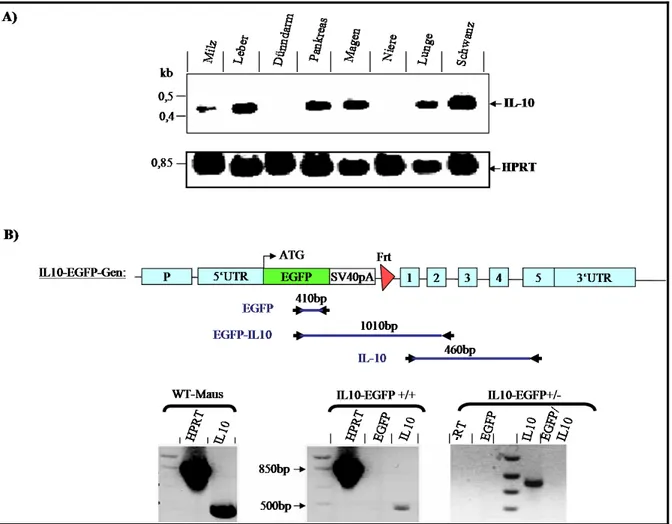
Etablierung von Interleukin-10 (IL-10)-Reportermäusen zur Identifizierung der IL-10-produzierenden Zellen in vivo
182
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
Abbildung

+7

ÄHNLICHE DOKUMENTE
In den Ergebnissen dieser Arbeit konnte kein Einfluss von CD80 oder CD86 auf die NK- Zell-Aktivierung durch sowohl TLR-DZ als auch cc-DZ nachgewiesen werden. Ob neben IL-12 noch
Interleukin-8 (IL-8) preferable to IL-6 as a marker for clinical infection. Clin Diagn Lab Immunol. Endothelial cell "memory" of inflammatory stimulation: human
Diese Ergebnisse zeigen, dass die Aktivierung von ERK1/2 für die Induktion der IL-10 Produk- tion auch in B-Zellen eine wichtige Rolle spielt und zur gesteigerten IL-10
Auch Kokulturen muriner Gefäßmuskelzellen, Fibroblasten oder Kardiomyozyten mit humanen mononukleären Zellen zeigten eine synergistische IL-6-Sekretion. Somit konnte
Neben ihrer Entstehung wurde auch die Funktion von IL10-produzierenden B-Zellen (B10) im Mausmodell untersucht. B10-Zellen sind in der Lage sowohl die Entstehung
Die Gene, die IL-1 α , IL-1 β und IL-1Ra kodieren, liegen beim Mensch in einem Genlocus auf dem Chromosom 2 und für einige Kombinationen dieser Gene wurde bereits
Neugeborene mit symptomatischer kongenitaler Toxoplasmose hatten signifikant erhöhte IL-18 Werte im Vergleich zu asymptomatischen Neugeborenen; lediglich tendenziell höhere IL-18 und
Weiterhin konnte mittels ELISA in T-Zell-Kokulturen mit PGN-stimulierten MoLC oder MoDC eine erhöhte Sekretion von IFN γ sowie IL-17, nicht jedoch von dem T
