Die Rolle der Ca /Calmodulin abhängigen Proteinkinase II in der akuten Phase der Nachlasterhöhung im Herzen
124
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
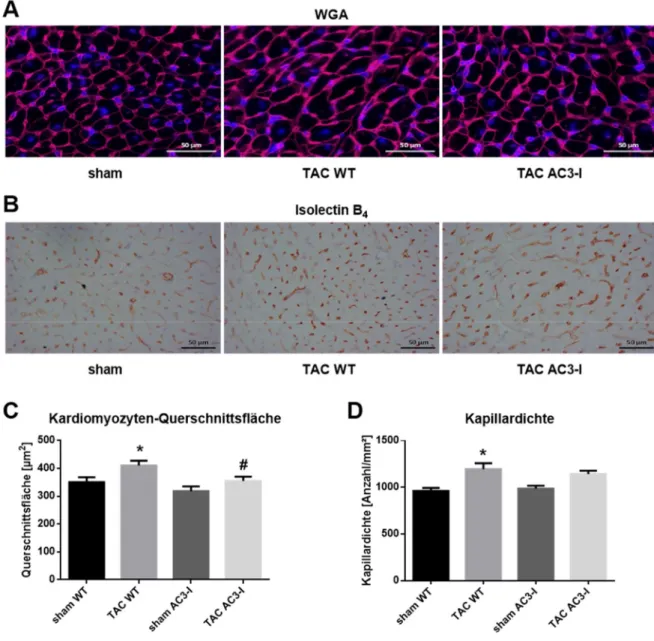
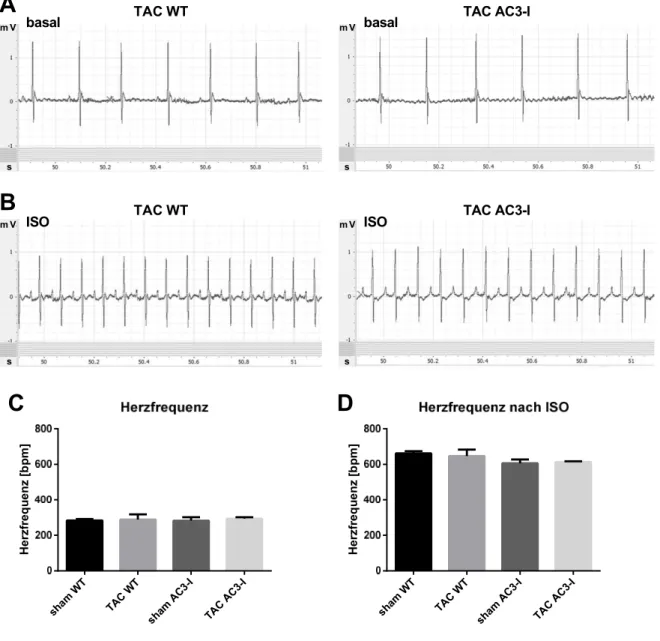
Abbildung
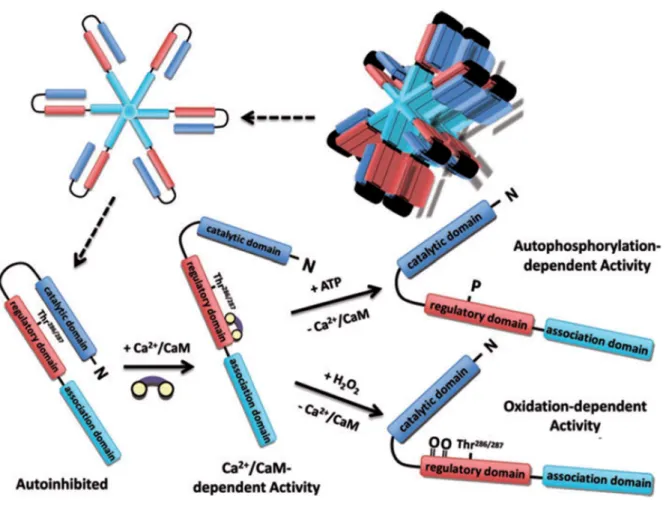
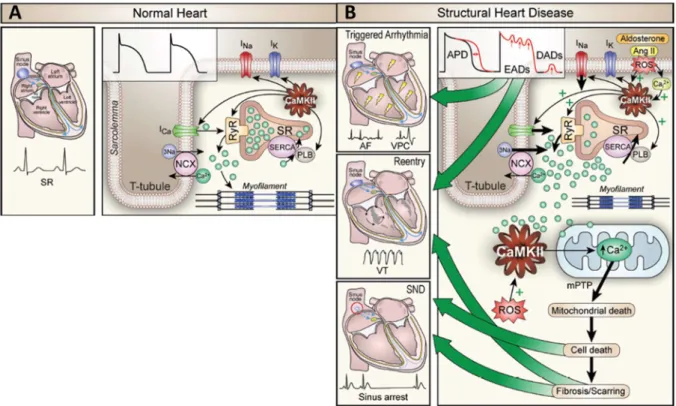
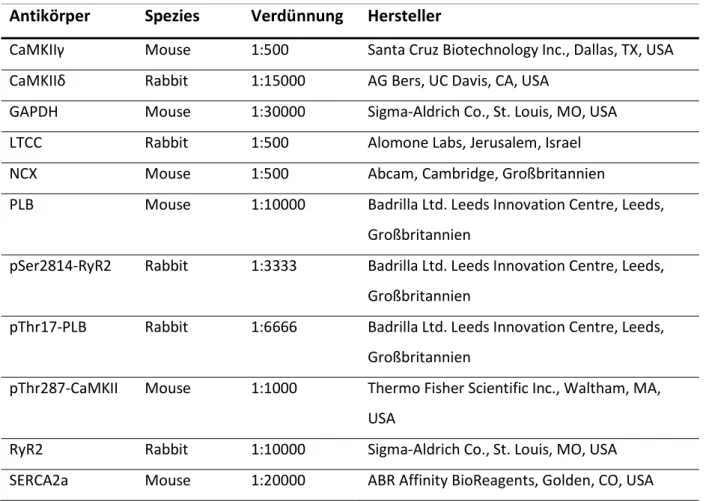
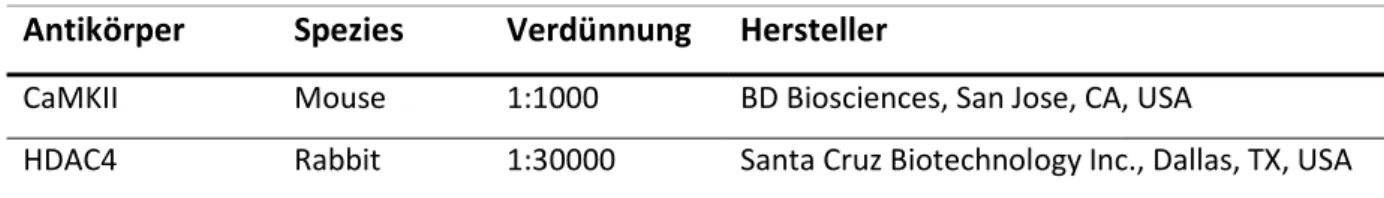
+7
ÄHNLICHE DOKUMENTE
- : Seasonal cycle experiments on the climate sensitivity due to a doubling of C02 with an atmospheric general circulation model coupled to a simple mixed
Ägyptens Kampagne auf dem Sinai etwa ist gerade in ihr viertes erfolg- loses Jahr getreten; im Jemen haben sich die Huthi-Rebellen zurückgemel- det, und das nach einer Dekade, in der
Analog konnte in dieser Dissertation gezeigt werden, dass ein erhöhtes SR-Ca 2+ -Leck im humanen terminal insuffizienten Myokard durch Hemmung der CaMKII mittels AIP reduziert
lichen Ausgaben sowie der grössere Teil der Mindereinnahmen wurden bereits früher im Jahr 2008 beschlossen und sind auf die gute Finanzlage der Kantone aufgrund der
Schlieÿlich konnte gezeigt werden, dass Sauerstoradikale über eine Ca 2+ -abhängige Aktivierung der CaMKIIδc eine Verstärkung des späten Na + - Stroms verursachen, was wiederum eine
Trotz der klaren Befunde und neuen Erkenntnisse über die vielfältige Rolle der PDE2 im Herzen lässt sich bisher noch nicht klar belegen, ob eine zusätzliche Aktivierung
Die in der Ausstellung Autofiktionen gezeigten Werke der für den Prix de dessin der Fondation d’art contemporain Daniel & Florence Guerlain ausgewählten Künstlerinnen und
Am d6 erfolgte eine weitere deutliche Steigerung der uPAR Expression im Kortex und im OSOM (Abb. Zusammenfassend konnte sowohl auf mRNA-Ebene als auch auf Proteinebene eine
