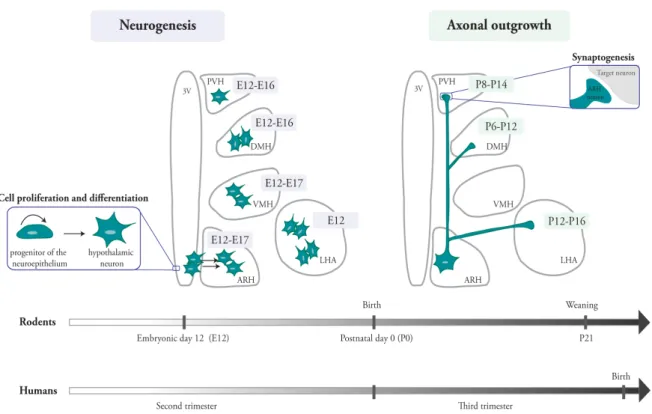
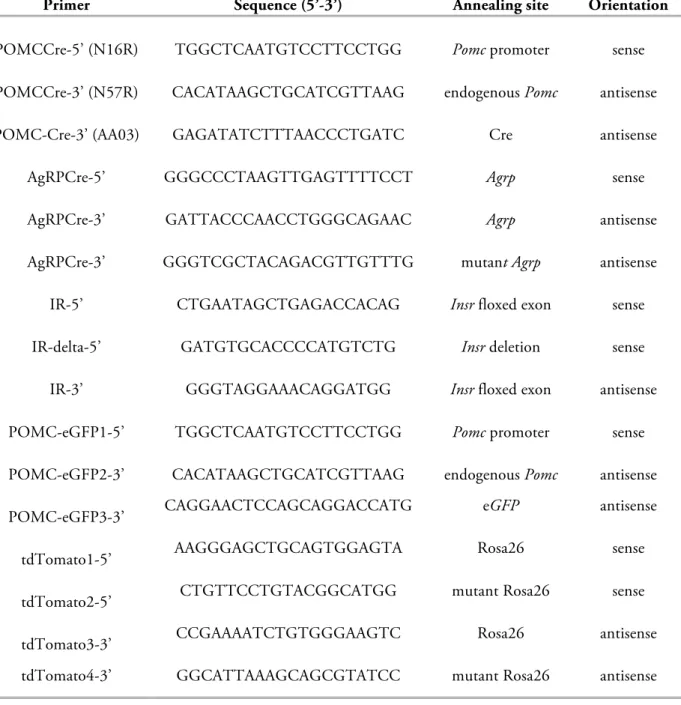
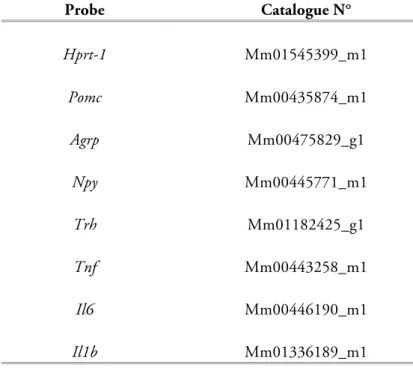
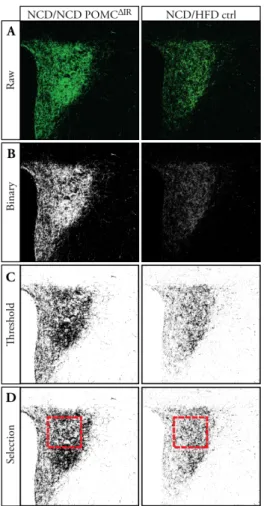
Metabolic Programming
135
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
Abbildung
+7
ÄHNLICHE DOKUMENTE
Constant hires - the condititon for a stable Beveridge curve - together with the common assumption of a fixed labor force (which allows for mobility between employment and
The intake of the fermented milk resulted in specific serum metabolomics profiles and was characterised by the modulation of metabolites of exogenous or endogenous origin, such as
fumigatus grown under normoxic or hypoxic conditions revealed an increased abundance of enzymes involved in the biosynthesis of secondary metabolites (Table 1 and Figure 1),
For instance, ethosux- imide is only effective in the GAERS model and almost exclusively used for the treatment of absence seizures in humans; phenytoin and carbamazepine act
Table 1 | Summary of typical examples of analysis methods and corresponding results produced with different modeling formalisms grouped in data types, which will be assigned
Welsh Water (Dŵr Cymru), Severn Trent Water, South West Water, Yorkshire Water and Anglian Water utilised paid social media promotion or geo-targeted posting to ensure they
The structure of brivaracetam was shown incorrectly (a hydroxyl group was shown instead of a keto group)2. In addition, the order of brivaracetam and levetiracetam was
The aim of my suggested approach (Möhner 2015) was to develop a method, which can even be applied to pub- lished results from cohort studies like the US coal miner mortality
