Research Collection
Doctoral Thesis
Evolutionary and population genomics of the wheat pathogen Parastagonospora nodorum
Author(s):
dos Santos Pereira, Danilo A.
Publication Date:
2020-09
Permanent Link:
https://doi.org/10.3929/ethz-b-000453101
Rights / License:
In Copyright - Non-Commercial Use Permitted
Evolutionary and population genomics of the wheat pathogen Parastagonospora nodorum
A thesis submitted to attain the degree of DOCTOR OF SCIENCES of ETH ZURICH
(Dr. sc. ETH Zurich)
presented by
DANILO AUGUSTO DOS SANTOS PEREIRA MSc São Paulo State University, Brazil
Born on 16.03.1989 Citizen of Brazil
accepted on the recommendation of Prof. Bruce McDonald
Prof. Daniel Croll Prof. Eva Stukenbrock
2020
This work is dedicated to the memory of Patrick Christoph Brunner.
You showed me that to be a good scientist one does not need to forget his family, friends, or hobbies. From my whole heart I will always be thankful for everything you taught me, particularly to be fast when there is cake around.
“Oh, I get by with a little help from my friends Mm, gonna try with a little help from my friends Oh, I get high with a little help from my friends”
John Lennon / Paul McCartney
SUMMARY ... 5
ZUSAMMENFASSUNG ... 7
GENERAL INTRODUCTION ... 9
Impact of plant pathogens in the food security ... 10
The challenge in the agro-ecosystem: a cradle for pathogen adaptation ... 11
Population genomics: a snapshot into the past, present, and future ... 12
A priori phenotype knowledge: phenotype-genotype association in plant pathogens .... 13
Posteriori phenotype knowledge: genome-wide selection scan in plant pathogens ... 15
The culprit: Parastagonospora nodorum biology, cycle, and management ... 16
Classical population genetics ... 17
The current status of genomics and evolutionary inferences in P. nodorum ... 18
The present PhD thesis ... 20
References ... 22
CHAPTER 1: Mutations in the CYP51 gene reduce DMI sensitivity in Parastagonospora nodorum populations in Europe and China ... 30
CHAPTER 2: Natural selection drives population divergence for local adaptation in a wheat pathogen ... 59
CHAPTER 3: The genetic architecture of emerging fungicide resistance in populations of a global wheat pathogen ... 106
CHAPTER 4: The population genomics of transposable element activation in the highly repressive genome of an agricultural pathogen ... 154
GENERAL DISCUSSION ... 210
Overview ... 211
Genetic diversity fueling a high adaptive potential in Parastagonospora nodorum populations ... 213 The genetic contributors to fungicide resistance variation in Parastagonospora
General conclusions and outlook ... 217
References ... 219
ACKNOWLEDGEMENTS ... 223
SCIENTIFIC CONTRIBUTIONS ... 225
CURRICULUM VITAE ... 227
Food production is continuously under threat by plant pathogens. In the agricultural system, pathogen populations are under high selection pressure, and yet demonstrate high adaptive potential to overcome changing conditions. The genetic variation and genomic mechanisms underpinning the fast evolutionary rates in pathogen populations are poorly understood. In this thesis, I studied the genetic basis of adaptation in populations of Parastagonospora nodorum, a major pathogen wheat. We sequenced complete genomes of 172 isolates sampled from important wheat growing regions worldwide, including Oregon, Texas, and New York (United States), Switzerland, Iran, South Africa, and Australia. These populations were sampled between 1991 and 2010 and incorporate a wide range of climatic conditions and agricultural practices. First, I tested the hypothesis of natural selection causing population divergence for local adaptation. I performed a common-garden experiment to collect precise quantitative phenotypic estimates of growth rates and melanization under a range of fungicide concentrations and temperatures. Phenotypic variation was used to estimate population differentiation for quantitative traits among populations (QST). The population genetic differentiation (FST) was calculated based on 50,000 neutral single nucleotide polymorphism (SNPs) markers. Employing a QST – FST comparison, I found that growth rates at 30°C and fungicide resistance estimates were under diversifying selection among populations while balancing selection was predominant for growth rates at 18°C and 24°C, as well for melanization. I found evidence for phenotypic trade-offs between growth rates and melanization at 30°C. Next, I investigated the genetic basis underlying variation for fungicide resistance. I show that non-synonymous mutations in the target gene CYP51 are linked to increased fungicide resistance against demethylation inhibitors. Furthermore, I performed a genome-wide association study to map phenotypic variation to a SNP dataset of 436,365
significantly associated SNPs, including CYP51 and a novel major facilitator superfamily (MFS) linked to fungicide variation. The increase in resistance as determined by the joint effects of the MFS and CYP51 mutations occurred in a non-additive manner. I found no evidence for genetic trade-offs constraining fungicide resistance evolution. Finally, I included additional genome sequences totalling 366 strains to resolve the population structure of P.
nodorum and investigate transposable elements (TEs) as drivers of genetic variation. I corroborate that populations are highly admixed, harbor high genetic diversity, and show evidence for frequent sexual recombination. Interestingly, despite robust genomic defenses called repeat induced-point (RIP) mutations, I found evidence for recently inserted TEs across the genome. I found no support that genetic bottlenecks incurred a TE burst among populations in P. nodorum. Moreover, TE copy number expansion was driven by miniature TEs as shown by a high GC content indicating an escape from RIP mutations. Overall, this PhD thesis demonstrates the potential of population genomics in unveiling evolutionary forces driving pathogen adaptation, as well as the genetic basis of adaptation and different sources of genetic novelty among populations.
Die Lebensmittelproduktion ist ständig durch Pflanzenpathogene bedroht. Im landwirtschaftlichen System stehen Pathogenpopulationen unter hohem Selektionsdruck und weisen dennoch ein hohes Anpassungspotential zur Überwindung sich ändernder Bedingungen auf. Die genetische Variation und die genomischen Mechanismen, die den schnellen Evolutionsraten in Pathogenpopulationen zugrunde liegen, sind kaum bekannt. In dieser Arbeit untersuchte ich die genetischen Grundlagen der Anpassung in Populationen von Parastagonospora nodorum, einem Hauptpathogen des Weizens. Wir sequenzierten die Genome von 172 Isolaten, die aus wichtigen Weizenanbaugebieten weltweit entnommen wurden, darunter Oregon, Texas und New York (USA), der Schweiz, dem Iran, Südafrika und Australien, vollsändig. Diese Populationen wurden zwischen 1991 und 2010 beprobt und umfassen eine breite Palette von klimatischen Bedingungen und landwirtschaftlichen Praktiken. Zuerst habe ich die Hypothese der natürlichen Selektion getestet, welche eine Populationsdivergenz zur lokalen Anpassung verursacht. Ich führte ein Common-Garden- Experiment durch, um genaue quantitative phänotypische Schätzungen der Wachstumsraten und der Melanisierung unter einer Reihe von Fungizidkonzentrationen und Temperaturen zu sammeln. Die phänotypische Variation wurde verwendet, um die Populationsdifferenzierung für quantitative Merkmale zwischen Populationen (QST) abzuschätzen. Die populationsgenetische Differenzierung (FST) wurde basierend auf 50.000 neutralen Einzelnukleotid-Polymorphismus (SNPs) -Markern berechnet. Unter Verwendung eines QST- FST-Vergleichs stellten wir fest, dass die Wachstumsraten bei 30 ° C und die Fungizidresistenzschätzungen zwischen den Populationen unter diversifizierender Selektion waren, während eine ausgleichende Selektion bei den Wachstumsraten bei 18 ° C und 24 ° C
genetische Basis, die der Variation der Fungizidresistenz zugrunde liegt. Ich zeige, dass nicht- synonyme Mutationen im Zielgen CYP51 mit einer erhöhten Fungizidresistenz gegen Demethylierungsinhibitoren verbunden sind. Darüber hinaus führte ich eine genomweite Assoziationsstudie durch, um die phänotypische Variation auf einen SNP-Datensatz von 436.365 genomweiten bi-allelischen Markern abzubilden, der neue Resistenzdeterminanten enthüllte. Wir fanden 34 signifikant assoziierte SNPs, einschließlich CYP51 und einer neuartigen Major Facilitator Superfamilie (MFS), die mit Fungizidvariationen verbunden sind.
Die durch die gemeinsame Wirkunge der MFS- und CYP51-Mutationen bestimmte Erhöhung der Resistenz erfolgte nicht additiv. Ich fand keine Hinweise auf genetische Kompromisse, die die Entwicklung der Fungizidresistenz behindern. Schließlich habe ich zusätzliche Genomsequenzen von insgesamt 366 Stämmen aufgenommen, um die Populationsstruktur von P. nodorum aufzuschlüsseln und transponierbare Elemente (TEs) als Treiber der genetischen Variation zu untersuchen. Ich bestätige, dass die Populationen stark vermischt sind, eine hohe genetische Vielfalt aufweisen und Hinweise auf eine häufige sexuelle Rekombination aufweisen. Interessanterweise fand ich trotz robuster genomischer Abwehrkräfte, die als RIP- Mutationen (Repeat Induced Point) bezeichnet werden, Hinweise auf kürzlich ins Genom eingefügte TEs. Ich fand keine Belege dafür, dass genetische Engpässe einen TE-Ausbruch unter den Populationen in P. nodorum verursachten. Darüber hinaus wurde die Erweiterung der TE-Kopienzahl durch Miniatur-TEs vorangetrieben, wie ein hoher GC-Gehalt zeigt, der auf ein Entkommen aus RIP-Mutationen hinweist. Insgesamt zeigt diese Doktorarbeit das Potenzial der Populationsgenomik, evolutionäre Kräfte aufzudecken, die die Anpassung von Krankheitserregern vorantreiben, sowie die genetischen Grundlagen der Anpassung und verschiedene Quellen genetischer Neuheit in Populationen.
General Introduction
Impact of plant pathogens in the food security
Despite technological advances since the advent of agriculture, humankind still faces the challenge of feeding the current population. An estimated over 820 million people cannot access the minimum food supply to conduct an active life (FAOSTAT, 2019). The situation is worsened under projections that the population might increase to 9.7 billion people by 2050 and about 11 billion by 2100 (Ganivet, 2019; United Nations, 2019). This situation of uncertainty on the fate of a growing population will affect the entire planet but will be aggravated in the poorest regions (Tilman et al., 2011; FAOSTAT, 2019). The physical availability of food is directly affected by the levels of food production, mainly from cereals, the largest source of calories for humankind (FAOSTAT, 2019; Savary et al., 2019). The major cereal producing areas are therefore pushed to increase crop yield or expand cropland area.
However, projections indicate a low yearly yield increase and high environmental costs for agricultural land expansion (Ray et al., 2013). Hence, an alternative solution is reducing losses in crop production to increase the availability of food in the supply chain. Significant losses in the agricultural ecosystem (agro-ecosystem) are related to changing environmental conditions and diseases caused by pests or pathogens (Chakraborty and Newton, 2011). Although seemingly unrelated, these are both interconnected problems. Plant pathogens cause major losses in crop productions, and with the aggravating climate crisis, the losses are predicted to increase (Strange and Scott, 2005; Chakraborty and Newton, 2011; Ganivet, 2019). At times this situation is worsened as evidenced in human history with famine (Padmanabhan, 1973;
Goodwin et al., 1994) and recent outbreaks (Islam et al., 2016; Almeida, 2018). Therefore, predicting disease outbreaks and designing efficient management strategies is necessary to prevent major yield losses in agriculture.
The challenge in the agro-ecosystem: a cradle for pathogen adaptation
The agro-ecosystems often imposes conducive environment for pathogen adaptation and the emergence of outbreaks (Burdon and Thrall, 2008). Mostly, this ecosystem had its components and landscape altered via anthropogenic actions to simplify and facilitate agricultural practices (McDonald and Linde, 2002). The most striking difference falls under the scope of diversity (Isbell et al., 2017). Compared to natural ecosystems, which are characterized by a high diversity of species per area, modern agro-ecosystems had a stark reduction in diversity, with densification of an individual clonal plant (McDonald and Linde, 2002). Such characteristics impose a strong directional selective pressure on pathogen populations, as pathogens must adapt and reproduce to transmit from host to host or go extinct. Besides adapting to the host, plant pathogens need to adapt to local environmental conditions (Laine, 2005). On the global scale, the agro-ecosystem is practiced under various conditions of environmental temperatures, rain regime, and daylight intensity. This joint of selective pressures adds further complexity to selection, which requires a versatile genetic structure for pathogen populations in order to adapt. The genetic structure is the concept of diversity in terms of genetic variation maintained within and among populations (McDonald and Linde, 2002). The genetic structure of a population is shaped according to the interplay of five evolutionary forces. (i) Mutations are the raw material for selection to act upon, as it alters the DNA and creates new alleles within populations (e.g., a new allele that confers fungicide resistance). (ii) The sexual reproductive system will recombine genotypes and put together new arrangements of alleles, which in combination with the asexual reproductive system, might multiply advantageous combinations (e.g., sexual and asexual reproductive cycles). (iii) gene flow or migration is the exchange of genetic material between or among populations geographically separated. (iv) Genetic drift is a ubiquitous evolutionary force causing stochastic changes in allele content, either increasing or decreasing frequencies (e.g., founder effect or bottlenecks). (v) Natural selection is the force
that will favor the fittest individual inserted in an environment, which consequently will increase its genetic combination in future generations. The presence of greater effective population sizes, continental gene flow, high genetic diversity, and the mixed reproductive system are characteristics found in several plant pathogen populations, rendering several species as rapidly adapting pathogens (McDonald and Linde, 2002). Therefore, investigating the evolutionary history of pathogen populations is essential to design efficient management strategies to disrupt their adaptability and reduce economic damage.
Population genomics: a snapshot into the past, present, and future
Population genomics has been defined as a new discipline that incorporates knowledge of classical evolutionary genetics to the resourcefulness of genomic tools (Black et al., 2001;
Luikart et al., 2003). In principle, genomics not only increases by many-fold the number of genetic markers, but it permits a detailed understanding of virtually any locus-specific and genome-wide effects beyond neutral inferences (Grünwald et al., 2016). Consequently, predictions about local adaptation can incorporate more broadly the complex interplay of gene flow, genetic drift, and natural selection in highly quantitative traits, connecting phenotypic variation with genetic variation (Savolainen et al., 2013).
For example, classical methods that compare genetic and phenotypic variation at the population level would profit from genomic advances. As the agro-ecosystem imposes strong biotic and abiotic selective pressure on pathogen populations, genes related to enabling host infection and persistence against environmental factors might be differently selected for local conditions (Stukenbrock and Bataillon, 2012). Comparisons of genetic variation for neutral markers (FST) and variation in quantitative traits (QST) allow differentiating between genetic drift (e.g.,
populations of fungal and oomycete plant pathogens have been studied in a QST – FST context where traits related to thermal adaptation, fungicide resistance, virulence, and melanization were found to be differently responding to selection among populations (Zhan et al., 2006;
Zhan and McDonald, 2011; Stefansson et al., 2014; Yang et al., 2016). These seminal studies paved the path for narrowed studies on specific genes related to pathogen life-history traits under selection (Zhong et al., 2017). Advanced genomic approaches that would allow identification of causal genetic variants (e.g., association mapping) or genomic loci (e.g., genome scans) possibly contributing to local adaptation could be further employed.
A priori phenotype knowledge: phenotype-genotype association in plant pathogens Correlating genetic variants to differences in phenotype is a long-standing quest in genetics.
The goal can be achieved by using classical quantitative trait locus (QTL) mapping or genome- wide association studies (GWAS; Grünwald et al., 2016). The difference between the two methods laydowns to the source of recombination incorporated (Myles et al., 2009). QTL mapping considers a cross-population of two parental organisms, often with contrasting phenotypes. The genome is divided into regions represented by genetic markers under linkage disequilibrium, and chromosomal regions can be associated with phenotypic variation.
Advantages of QTL mapping include its resourcefulness to unveil rare alleles contributing to the phenotypic variation and that it is not affected by population structure (Bergelson and Roux, 2010). With a GWAS, the genetic variation obtained is sourced from natural (e.g., field) populations and can represent a full phenotype spectrum. Advantages of a GWAS include the potential to exploit all the effects from genetic recombination from the evolutionary history of a sample, and not only segregating sites. The mapping resolution is also higher in a GWAS.
GWAS is particularly suitable for the organisms in which populations experience significant gene flow, undergoes frequent sexual recombination, and are highly genetically diverse (Korte
and Farlow, 2013). Moreover, GWAS becomes even more remarkable when sexual crosses in laboratory conditions are not feasible. Furthermore, GWAS methods that control for false positives and false negatives are necessary to circumcise confounding effects from population structure and cryptic relationship. An efficient method to control for spurious associations is the mixed linear models (MLM), including a matrix of kingship scores (Price et al., 2010;
Sillanpää, 2011).
Given an adequate whole-genome sequencing with sufficient coverage, the success of GWAS primarily depends on the sample size and accuracy of scoring phenotypes (Myles et al., 2009).
Small differences in phenotype variation might be challenging to measure but are adequate for selection to act upon. Initial GWAS in plant pathogens focused on identifying virulence factors in either a reduced panel of isolates (Dalman et al., 2013) or reduced marker density (Gao et al., 2016; Talas et al., 2016). Later, these constraints were overcome with successful identification and characterization of candidate genes (Hartmann et al., 2017; Zhong et al., 2017). Interestingly, the avirulence gene AvrStb6 was found in both a GWAS and QTL mapping, highlighting the robust complementarity of these methods. Despite previous GWAS applications in plant pathogen, the focus has been kept in investigating pathogenicity determinants (Bartoli and Roux, 2017). The potential of GWAS to unveil fungicide resistant determinants has been recently demonstrated in the barley – Rhynchosporium commune pathosystem (Mohd-Assaad et al. 2016). A single nucleotide polymorphism (SNP) marker dataset was mapped to phenotypic variation against an Azole fungicide, and novel mechanisms related to increased resistance were postulated. Additionally, recent development in scoring phenotypes using automated image analysis has improved the probability of finding causal genetic variants through GWAS (Stewart and McDonald, 2007; Lendenmann et al., 2014).
Combining next-generation sequencing with precise and automated high throughput phenotype acquisition holds the potential to unveil the genetic basis of complex traits.
Posteriori phenotype knowledge: genome-wide selection scan in plant pathogens
Population genomics enabled the use of genome scans to identify genomic regions under positive selection. While QST-FST comparisons aim to unveil if a trait is under selection for local adaptation and GWAS is powerful to pinpoint the candidate locus responsible for variation in the trait, they are both grounded on phenotypic information a priori. Selection scans leverage of a different approach for inferences on the action of natural selection along the genome (Vitti et al., 2013). Instead of choosing a putative adaptative trait and tracking its genetic polymorphism, it is possible to inversely infer the phenotypic functions beginning from the genetic information (termed reverse genetics; Li et al., 2008). The rationale relies on the theory that selective sweeps will represent the increase in the frequency of a beneficial mutation with a reduction in genetic diversity in its surroundings (e.g., compared to the genetic background; Vitti et al., 2013; Grünwald et al., 2016). Depending on the source of the advantageous mutation, the reduction in diversity changes. Soft sweeps (or incomplete sweeps) are expected to source from genetic variation in the standing genetic variation of a population (Hermisson and Pennings, 2005), while hard sweeps are considered to be originated from rare mutations (Maynard and Haigh, 2007) throughout the genome. Further analysis of the gene content in selective weeps would give hints on gene function and role. Applying the reverse genetics approach in plant pathogens was possible to identify the genes associated to host specialization and adaptation to agriculture important conditions (Stukenbrock et al., 2011;
Brunner et al., 2013; Badouin et al., 2017). Further studies identified genes associated to environmental adaptation and virulence (Hartmann et al., 2018; Mohd-Assaad et al., 2018).
These examples highlight the use of genomic tools to explore the different facets of pathogen evolution.
The culprit: Parastagonospora nodorum biology, cycle, and management
The haploid ascomycete Parastagosnopora nodoroum (formally anamorph: Stagonospora nodorum, and teleomorph: Phaeosphaeria nodorum) is the causal disease agent of Stagonospora nodorum botch (SNB), a major disease of common and durum wheat, barley (Osbourn et al., 1986; Quaedvlieg et al., 2013), and secondarily found in wild grasses (Williams and Jones, 1973). This fungal pathogen damages the head and leaves of wheat during the cropping season, decreasing both grain quality and yield (Bhathal et al., 2003). On the leaf surface, infection occurs via natural openings (e.g., stomata) or wounds, but direct penetration through the cuticle is the main route (Solomon et al., 2006). Different from other foliar blotch diseases, infection of P. nodorum in a susceptible host under conducive conditions renders lesions within a few days, and a complete asexual cycle is fulfilled in less than seven days (Solomon et al., 2006). At the initial phase of host colonization, visible chlorotic patches are contrasted with the natural green color of a healthy leaf, which indicates the fungal penetration site. As the colonization progresses, chlorotic blotches develop into larger oval-shaped lesions, with a brownish center and signs of a chlorotic surrounding. In the late phase of colonization, asexual fruiting bodies called pycnidia start to form within the lesions (Solomon et al., 2006).
The pycnidia primordium is not distinguishable from the lesion background, but soon darken and become brown (Douaiher et al., 2004). Mature pycnidia contains millions of spores called pycnidiospores that will give rise to new secondary infection cycles within the growing season.
The sexual cycle has also been described, and the sexual spores (ascospores) mainly overwinter in stubbles, which play important role in long-distant dispersal (Bathgate and Loughman, 2001;
of this pathogen (Shah, 1995). Control methods for SNB rely mostly on the deployment of resistant hosts (e.g., wheat varieties lacking susceptibility genes) and chemical treatments (e.g., spraying of fungicides; Ficke et al., 2017).
Modern fungicides used in chemical disease management are usually site-specific and have higher control efficiency (Deising et al., 2008). However, upon constant and intensive applications, fungicides exert high selective pressure over pathogen populations, and individuals with lower susceptibility quickly increase in frequency (McDonald and Linde, 2002). For example, like many other plant plant pathogens, mutation in the mitochondrial- encoded cytochrome b gene conferring resistance against strobilurins in P. nodorum is common in field conditions (Blixt et al., 2009). Gradually, the chemical control of SNB and other co- occurring diseases of wheat relies more on demethylation inhibitor (DMI) fungicides (e.g., azoles), one of the most used fungicide class in Europe (Blixt et al., 2010; Torriani et al., 2015).
However, as azoles are also site-specific fungicides (i.e., target the CYP51 gene), the emergence of mutations correlated to a reduction in susceptibility is expected to surface in P. nodorum despite recent survey (Blixt et al., 2009).
Classical population genetics
The first studies on population genetics of P. nodorum employed different molecular approaches to unveil the levels of diversity in the species. Using protein electrophoresis patterns (Durbin, 1966) to restriction fragment length polymorphism (McDonald, 1994; Keller et al., 1997) and microsatellites makers (Stukenbrock, Banke, and McDonald, 2006), presence of high genetic variation in this pathogen was confirmed. For example, in single fields, high degrees of diversity were found widespread, with an average of more than 95% of the total gene diversity found locally. Interestingly, different haplotypes were present in a single infected wheat leaf
(Keller et al., 1997). The clonality was generally low (≤ 16%) among populations, and important routes of gene flow were established among continents (Stukenbrock, Banke, and McDonald, 2006). The pathogen was later postulated to have emerged in the ancient Fertile Crescent, given the presence of closely related grass species and a local hotspot of diversity in an Iranian population (McDonald et al., 2012). It has been recently shown that diversity varies among Iranian populations, possibly due to the levels of population subdivision within the country (Ghaderi et al., 2020). This hypothesis of the center of origin is consistent with the considered birthplace of human agriculture and wheat domestication bout 12’000 years ago (Braidwood et al., 1969; Charmet, 2011; McDonald et al., 2012; Lazaridis et al., 2016; Ghaderi et al., 2020). Via host-tracking of wheat, P. nodorum was dispersed and introduced into Europe, Africa, Asia, and the Americas (Salamini et al., 2002). Since its spread, SNB is now common in wheat growing-regions, causing significant yield losses (Ficke et al., 2017). Given the global presence of P. nodorum, more information on the genetic basis of adaptation is necessary to properly tackle its evolutionary potential.
The current status of genomics and evolutionary inferences in P. nodorum
Since the first genome sequence published for a plant pathogen (Simpson et al., 2000), it has established a new perspective for studies on the genetics of these organisms. In a matter of time, there was a broad expansion in the availability of genome sequences for various species, allowing gaps to be filled between fundamental and applied research areas. The whole-genome sequence for P. nodorum reference strain SN15 was published in 2007 (Hane et al., 2007), followed by re-annotation and improvements in 2016 (Syme et al., 2016). Genome sequences of nearly complete telomere-to-telomere genomes were later published for other reference strains (Richards et al., 2019), and a pangenome assembled (Syme et al., 2018). Electrophoretic
strains having similar migration bands (Neil Cooley and Caten, 1991). Later, based on whole- genome sequencing, the smallest chromosome (number 23) was identified as accessory or dispensable (Ohm et al., 2012; Bertazzoni et al., 2018). The nuclear genome of P. nodorum is about 37.1Mb, comprising approximately 4,5% of repetitive DNA sequence for reference SN15 (Hane et al., 2007). The genomic activity of transposable elements (TEs) is largely assumed to be controlled by repetitive induced point (RIP) mutations in P. nodorum. RIP is a genome mechanism that targets repetitive sequences during meiosis, and various families of TEs have been found to hold evidences of RIP in this pathogen (Hane and Oliver, 2008; Oliver et al., 2012).
These valuable resources have leveraged P. nodorum as a model organism among necrotrophic pathogens for studies on necrotrophic effectors (NEs; Liu et al., 2004; Oliver et al., 2012). The NEs interact with dominant susceptibility genes in wheat, and in an inverse gene-for-gene manner (Flor, 1956), it subverts the normal wheat resistance response to exploit the resulting necrosis (Friesen et al., 2007). Resistance against SNB is considered to be broadly quantitative (e.g., polygenic; Oliver et al., 2012), but with significant contributions from NE genes and susceptibility genes interactions. Overall, a total of three pathogen NEs have been characterized and cloned (SnTox1, SnToxA, SnTox3), and nine other interactions reported (Friesen et al.
2007, 2012; Gao et al. 2015). NEs were also found to be under selective pressure imposed by the host (Stukenbrock and Mcdonald, 2007; Liu et al., 2012), display presence-absence variation (Stukenbrock and Mcdonald, 2007) and to be located near TEs (Friesen et al., 2006;
Richards et al., 2019). In fact, such dynamism for NEs are also indicators of an overlooked evolving genomic landscape. For example, P. nodorum stands as one of the few filamentous plant pathogens to have been characterized for the occurrence of a horizontal gene transfer (HGT; Friesen et al., 2006). The ToxA gene, initially identified in Pyrenophora tritici-repentis,
was found to source from P. nodorum (Friesen et al., 2006). This important gene is embedded in a TE rich region, which possibly increases the genomic dynamism in this region facilitating genetic changes (McDonald et al., 2018; McDonald et al., 2019). Despite the successful use and application of genomics in the P. nodorum wheat pathosystem, it has not surpassed beyond studies with a focus on NE. The genetic basis for variation in life-history traits in P. nodorum remains poorly understood. Further questions related to the genetic basis of adaptation towards environmental conditions and genetic differences among global populations at the whole- genome scale remains unanswered.
The present PhD thesis
The present work focuses on improving the understanding of the evolutionary genetic potential of P. nodorum employing population and comparative genomics tools. In the light of knowledge about evolutionary history, long-term management strategies might be designed.
Chapter 1 describes the first mutation in an important protein-coding gene targeted by modern Azole fungicides in P. nodorum. This novel mutation is demonstrated to be associated to higher resistance against a fungicide and worldwide spread.
Chapter 2 demonstrates the resulting action of selection acting towards local adaptation among populations of P. nodorum. I show that differences among populations for important agronomical traits are under different kinds of selection.
Chapter 3 dissects the genetic architecture of fungicide resistance in P. nodorum. I performed a combination of GWAS and correlation analysis to demonstrate the contribution of different loci to variation in fungicide resistance and possible trade-offs.
Chapter 4 details the evolutionary history of P. nodorum in the context of population structure with an expanded isolate collection. I describe global levels of population diversity, recent transposable elements activity, and distribution of selective sweeps across the genome.
References
Almeida RPP (2018) Emerging plant disease epidemics: biological research is key but not enough. PLoS Biol 16:e2007020.
Badouin H, Gladieux P, Gouzy J, Siguenza S, Aguileta G, Snirc A, Le Prieur S, Jeziorski C, Branca A, and Giraud T (2017) Widespread selective sweeps throughout the genome of model plant pathogenic fungi and identification of effector candidates. Mol Ecol 26:2041–2062.
Bartoli C, and Roux F (2017) Genome-wide association studies in plant pathosystems: toward an ecological genomics approach. Front Plant Sci 8:763.
Bathgate JA, and Loughman R (2001) Ascospores are a source of inoculum of Phaeosphaeria nodorum, P. avenaria f. sp. avenaria and Mycosphaerella graminicola in Western Australia. Austral Plant Pathol 30:317.
Bergelson J, and Roux F (2010) Towards identifying genes underlying ecologically relevant traits in Arabidopsis thaliana. Nat Rev Genet 11:867–879.
Bertazzoni S, Williams AH, Jones DA, Syme RA, Tan K-C, and Hane JK (2018) Accessories make the outfit: accessory chromosomes and other dispensable DNA regions in plant- pathogenic fungi. Mol Plant Microbe Interact 31:779–788.
Bhathal JS, Loughman R, and Speijers J (2003) Yield reduction in wheat in relation to leaf disease from yellow (tan) spot and Septoria nodorum Blotch. Eur J Plant Pathol 109:435–443.
Black WC, Baer CF, Antolin MF, and DuTeau NM (2001) Population genomics: genome-wide sampling of insect populations. Annu Rev Entomol 46:441–469.
Blixt E, Djurle A, Yuen J, and Olson Å (2009) Fungicide sensitivity in Swedish isolates of Phaeosphaeria nodorum. Plant Pathol 58:655–664.
Blixt E, Olson Å, Lindahl B, Djurle A, and Yuen J (2010) Spatiotemporal variation in the fungal community associated with wheat leaves showing symptoms similar to stagonospora nodorum blotch. Eur J Plant Pathol 126:373–386.
Braidwood RJ, Çambel H, and Watson PJ (1969) Prehistoric investigations in southeastern Turkey. Science 164:1275.
Brunner PC, Torriani SFF, Croll D, Stukenbrock EH, and McDonald BA (2013) Coevolution and life cycle specialization of plant cell wall degrading enzymes in a hemibiotrophic pathogen. Mol Biol Evol 30:1337–1347.
Burdon JJ, and Thrall PH (2008) Pathogen evolution across the agro-ecological interface:
Chakraborty S, and Newton AC (2011) Climate change, plant diseases and food security: an overview: Climate change and food security. Plant Pathol 60:2–14.
Charmet G (2011) Wheat domestication: Lessons for the future. CR Biol 334:212–220.
Dalman K, Himmelstrand K, Olson Å, Lind M, Brandström-Durling M, and Stenlid J (2013) A genome-wide association study identifies genomic regions for virulence in the non- model organism Heterobasidion annosum s.s. PLOS ONE 8:e53525.
Deising HB, Reimann S, and Pascholati SF (2008) Mechanisms and significance of fungicide resistance. Braz J Microbiol 39:286–295.
Douaiher MN, Halama P, and Janex-Favre MC (2004) The ontogeny of Stagonospora nodorum pycnidia in culture. Sydowia 56:39–50.
Durbin RD (1966) Comparative gel-electrophoretic investigation of the protein patterns of Septoria species. Nature 210:1186–1187.
FAOSTAT (2019) Food and Agriculture Organization of the United Nations.
Ficke A, Cowger C, Bergstrom GC, and Brodal G (2017) Understanding yield loss and pathogen biology to improve disease management: Septoria nodorum blotch - a case study in wheat. Plant Dis 102:696–707.
Flor HH (1956) The complementary genic systems in flax and flax rust, in Advances in Genetics pp 29–54, Elsevier.
Friesen T l., Chu C, Xu SS, and Faris JD (2012) SnTox5- Snn5 : a novel Stagonospora nodorum effector-wheat gene interaction and its relationship with the SnToxA- Tsn1 and SnTox3- Snn3 - B1 interactions: Characterization of the SnTox5- Snn5 interaction. Mol Plant Pathol 13:1101–1109.
Friesen TL, Meinhardt SW, and Faris JD (2007) The Stagonospora nodorum‐wheat pathosystem involves multiple proteinaceous host‐selective toxins and corresponding host sensitivity genes that interact in an inverse gene‐for‐gene manner. The Plant Journal 51:681–692.
Friesen TL, Stukenbrock EH, Liu Z, Meinhardt S, Ling H, Faris JD, Rasmussen JB, Solomon PS, McDonald BA, and Oliver RP (2006) Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet 38:953–956.
Ganivet E (2019) Growth in human population and consumption both need to be addressed to reach an ecologically sustainable future. Environ Dev Sustain, doi: 10.1007/s10668- 019-00446-w.
Gao Y, Faris JD, Liu Z, Kim YM, Syme RA, Oliver RP, Xu SS, and Friesen TL (2015) Identification and characterization of the SnTox6- Snn6 interaction in the Parastagonospora nodorum –wheat pathosystem. Mol Plant Microbe Interact 28:615–
625.
Gao Y, Liu Z, Faris JD, Richards J, Brueggeman RS, Li X, Oliver RP, McDonald BA, and Friesen TL (2016) Validation of genome-wide association studies as a tool to identify virulence factors in Parastagonospora nodorum. Phytopathology 106:1177–1185.
Ghaderi F, Sharifnabi B, Javan-Nikkhah M, Brunner PC, and McDonald BA (2020) SnToxA, SnTox1 and SnTox3 originated in Parastagonospora nodorum in the Fertile Crescent, bioRxiv.
Goodwin SB, Cohen BA, and Fry WE (1994) Panglobal distribution of a single clonal lineage of the Irish potato famine fungus. Proc Natl Acad Sci USA 91:11591–11595.
Grünwald NJ, McDonald BA, and Milgroom MG (2016) Population genomics of fungal and oomycete pathogens. Annu Rev Phytopathol 54:323–346.
Hane JK, Lowe RGT, Solomon PS, Tan K-C, Schoch CL, Spatafora JW, Crous PW, Kodira C, Birren BW, Galagan JE, Torriani SFF, McDonald BA, and Oliver RP (2007) Dothideomycete–plant interactions illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum. Plant Cell 19:3347–3368.
Hane JK, and Oliver RP (2008) RIPCAL: a tool for alignment-based analysis of repeat-induced point mutations in fungal genomic sequences. BMC Bioinform 9:478.
Hartmann FE, McDonald BA, and Croll D (2018) Genome‐wide evidence for divergent selection between populations of a major agricultural pathogen. Mol Ecol 27:2725–
2741.
Hartmann FE, Sánchez-Vallet A, McDonald BA, and Croll D (2017) A fungal wheat pathogen evolved host specialization by extensive chromosomal rearrangements. ISME J 11:1189–1204.
Hermisson J, and Pennings PS (2005) Soft Sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169:2335–2352.
Isbell F, Adler PR, Eisenhauer N, Fornara D, Kimmel K, Kremen C, Letourneau DK, Liebman M, Polley HW, Quijas S, and Scherer-Lorenzen M (2017) Benefits of increasing plant diversity in sustainable agroecosystems. J Ecol 105:871–879.
Islam MT, Croll D, Gladieux P, Soanes DM, Persoons A, Bhattacharjee P, Hossain MdS, Gupta DR, Rahman MdM, Mahboob MG, Cook N, Salam MU, Surovy MZ, Sancho VB, Maciel JLN, NhaniJúnior A, Castroagudín VL, Reges JT de A, Ceresini PC, Ravel S, Kellner R, Fournier E, Tharreau D, Lebrun M-H, McDonald BA, Stitt T, Swan D, Talbot NJ, Saunders DGO, Win J, and Kamoun S (2016) Emergence of wheat blast in Bangladesh was caused by a South American lineage of Magnaporthe oryzae. BMC Biol 14:84.
Keller S, McDermott J, Pettway R, Wolfe M, and McDonald B (1997) Gene flow and sexual reproduction in the wheat glume blotch pathogen Phaeosphaeria nodorum (anamorph Stagonospora nodorum). Phytopathology 87:353–358.
Korte A, and Farlow A (2013) The advantages and limitations of trait analysis with GWAS: a
Laine A-L (2005) Spatial scale of local adaptation in a plant-pathogen metapopulation. J Evolution Biol 18:930–938.
Lazaridis I, Nadel D, Rollefson G, Merrett DC, Rohland N, Mallick S, Fernandes D, Novak M, Gamarra B, Sirak K, Connell S, Stewardson K, Harney E, Fu Q, Gonzalez-Fortes G, Jones ER, Roodenberg SA, Lengyel G, Bocquentin F, Gasparian B, Monge JM, Gregg M, Eshed V, Mizrahi A-S, Meiklejohn C, Gerritsen F, Bejenaru L, Blüher M, Campbell A, Cavalleri G, Comas D, Froguel P, Gilbert E, Kerr SM, Kovacs P, Krause J, McGettigan D, Merrigan M, Merriwether DA, O’Reilly S, Richards MB, Semino O, Shamoon-Pour M, Stefanescu G, Stumvoll M, Tönjes A, Torroni A, Wilson JF, Yengo L, Hovhannisyan NA, Patterson N, Pinhasi R, and Reich D (2016) Genomic insights into the origin of farming in the ancient Near East. Nature 536:419–424.
Lendenmann MH, Croll D, Stewart EL, and McDonald BA (2014) Quantitative trait locus mapping of melanization in the plant pathogenic fungus Zymoseptoria tritici. G3 4:2519–33.
Li YF, Costello JC, Holloway AK, and Hahn MW (2008) “Reverse ecology” and the power of population genomics. Evolution 62:2984–2994.
Liu Z, Zhang Z, Faris JD, Oliver RP, Syme R, McDonald MC, McDonald BA, Solomon PS, Lu S, Shelver WL, Xu S, and Friesen TL (2012) The cysteine rich necrotrophic effector SnTox1 produced by Stagonospora nodorum triggers susceptibility of wheat lines harboring Snn1. PLOS Pathog 8:e1002467.
Liu ZH, Faris JD, Meinhardt SW, Ali S, Rasmussen JB, and Friesen TL (2004) Genetic and physical mapping of a gene conditioning sensitivity in wheat to a partially purified host- selective toxin produced by Stagonospora nodorum. Phytopathology 94:1056–1060.
Luikart G, England PR, Tallmon D, Jordan S, and Taberlet P (2003) The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet 4:981–994.
Maynard J, and Haigh J (2007) The hitch-hiking effect of a favourable gene. Genet Res 89:391–
403.
McDonald BA (1994) Genetic variability in nuclear DNA in field populations of Stagonospora nodorum. Phytopathology 84:250.
McDonald BA, and Linde C (2002) Pathogen population genetics, evolutionary potential, and durable resistance. Annu Rev Phytopathol 40:349–379.
McDonald MC, Ahren D, Simpfendorfer S, Milgate A, and Solomon PS (2018) The discovery of the virulence gene ToxA in the wheat and barley pathogen Bipolaris sorokiniana:
Discovery of the virulence gene ToxA. Mol Plant Pathol 19:432–439.
McDonald MC, Razavi M, Friesen TL, Brunner PC, and McDonald BA (2012) Phylogenetic and population genetic analyses of Phaeosphaeria nodorum and its close relatives indicate cryptic species and an origin in the Fertile Crescent. Fungal Genet Biol 49:882–895.
McDonald MC, Taranto AP, Hill E, Schwessinger B, Liu Z, Simpfendorfer S, Milgate A, and Solomon PS (2019) Transposon-mediated horizontal transfer of the host-specific virulence protein ToxA between three fungal wheat pathogens. mBio 10:e01515-19.
Mohd-Assaad N, McDonald BA, and Croll D (2018) Genome-wide detection of genes under positive selection in worldwide populations of the barley scald pathogen. Genome Biol Evol 10:evy087-.
Myles S, Peiffer J, Brown PJ, Ersoz ES, Zhang Z, Costich DE, and Buckler ES (2009) Association mapping: critical considerations shift from genotyping to experimental Design. Plant Cell 21:2194–2202.
Neil Cooley R, and Caten CE (1991) Variation in electrophoretic karyotype between strains of Septoria nodorum. Molec Gen Genet 228:17–23.
Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC, Dhillon B, Glaser F, Hesse CN, Kosti I, LaButti K, Lindquist EA, Lucas S, Salamov AA, Bradshaw RE, Ciuffetti L, Hamelin RC, Kema GHJ, Lawrence C, Scott JA, Spatafora JW, Turgeon BG, de Wit PJGM, Zhong S, Goodwin SB, and Grigoriev IV (2012) Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes Fungi. PLoS Pathog 8:e1003037.
Oliver R, Friesen T, Faris J, and Solomon P (2012) Stagonospora nodorum: from pathology to genomics and host resistance. Annu Rev Phytopathol 50:23–43.
Osbourn AE, Scott PR, and Caten CE (1986) The effects of host passaging on the adaptation of Septoria nodorum to wheat or barley. Plant Pathology 35:135–145.
Padmanabhan SY (1973) The great bengal famine. Annu Rev Phytopathol 11:11–24.
Price AL, Zaitlen NA, Reich D, and Patterson N (2010) New approaches to population stratification in genome-wide association studies. Nat Rev Genet 11:459–463.
Quaedvlieg W, Verkley GJM, Shin H-D, Barreto RW, Alfenas AC, Swart WJ, Groenewald JZ, and Crous PW (2013) Sizing up Septoria. Studies in Mycology 75:307–390.
Ray DK, Mueller ND, West PC, and Foley JA (2013) Yield trends are insufficient to double global crop production by 2050. PLOS ONE 8:e66428.
Richards J, Stukenbrock E, Carpenter J, Liu Z, Cowger C, Faris J, and Friesen TL (2019) Local adaptation drives the diversification of effectors in the fungal wheat pathogen Parastagonospora nodorum in the United States. PLoS Genet 15:393–399.
Salamini F, Özkan H, Brandolini A, Schäfer-Pregl R, and Martin W (2002) Genetics and geography of wild cereal domestication in the near east. Nat Rev Genet 3:429–441.
Savary S, Willocquet L, Pethybridge S, Esker P, McRoberts N, and Nelson A (2019) The global burden of pathogens and pests on major food crops. Nat Ecol Evol 3:430–439.
Savolainen O, Lascoux M, and Merilä J (2013) Ecological genomics of local adaptation. Nat
Shah D (1995) Initiation of Septoria nodorum blotch epidemics in winter wheat by seedborne Stagonospora nodorum. Phytopathology 85:452.
Sillanpää MJ (2011) Overview of techniques to account for confounding due to population stratification and cryptic relatedness in genomic data association analyses. Heredity 106:511–519.
Simpson AJG, Reinach FC, Arruda P, Abreu FA, Acencio M, Alvarenga R, Alves LMC, Araya JE, Baia GS, Baptista CS, Barros MH, Bonaccorsi ED, Bordin S, Bové JM, Briones MRS, Bueno MRP, Camargo AA, Camargo LEA, Carraro DM, Carrer H, Colauto NB, Colombo C, Costa FF, Costa MCR, Costa-Neto CM, Coutinho LL, Cristofani M, Dias- Neto E, Docena C, El-Dorry H, Facincani AP, Ferreira AJS, Ferreira VCA, Ferro JA, Fraga JS, França SC, Franco MC, Frohme M, Furlan LR, Garnier M, Goldman GH, Goldman MHS, Gomes SL, Gruber A, Ho PL, Hoheisel JD, Junqueira ML, Kemper EL, Kitajima JP, Krieger JE, Kuramae EE, Laigret F, Lambais MR, Leite LCC, Lemos EGM, Lemos MVF, Lopes SA, Lopes CR, Machado JA, Machado MA, Madeira AMBN, Madeira HMF, Marino CL, Marques MV, Martins EAL, Martins EMF, Matsukuma AY, Menck CFM, Miracca EC, Miyaki CY, Monteiro-Vitorello CB, Moon DH, Nagai MA, Nascimento ALTO, Netto LES, Nhani A, Nobrega FG, Nunes LR, Oliveira MA, de Oliveira MC, de Oliveira RC, Palmieri DA, Paris A, Peixoto BR, Pereira GAG, Pereira HA, Pesquero JB, Quaggio RB, Roberto PG, Rodrigues V, de M.
Rosa AJ, de Rosa VE, de Sá RG, Santelli RV, Sawasaki HE, da Silva ACR, da Silva AM, da Silva FR, et al. (2000) The genome sequence of the plant pathogen Xylella fastidiosa. Nature 406:151–157.
Solomon PS, Lowe RG, Tan K-C, Waters OD, and Oliver RP (2006) Stagonospora nodorum:
cause of stagonospora nodorum blotch of wheat. Mol Plant Pathol 7:147–156.
Stefansson TS, McDonald BA, and Willi Y (2014) The influence of genetic drift and selection on quantitative traits in a plant pathogenic fungus. PLOS ONE 9:e112523.
Stewart EL, and McDonald BA (2014) Measuring quantitative virulence in the wheat Pathogen Zymoseptoria tritici using high-throughput automated image analysis. Phytopathology 104:985–92.
Strange RN, and Scott PR (2005) Plant Disease: a threat to global food security. Annu Rev Phytopathol 43:83–116.
Stukenbrock EH, Banke S, and McDonald BA (2006) Global migration patterns in the fungal wheat pathogen Phaeosphaeria nodorum. Mol Ecol 15:2895–2904.
Stukenbrock EH, and Bataillon T (2012) A population genomics perspective on the emergence and adaptation of new plant pathogens in agro-ecosystems. PLoS Pathog 8:e1002893.
Stukenbrock EH, Bataillon T, Dutheil JY, Hansen TT, Li R, Zala M, McDonald BA, Wang J, and Schierup MH (2011) The making of a new pathogen: Insights from comparative population genomics of the domesticated wheat pathogen Mycosphaerella graminicola and its wild sister species. Genome Res 21:2157–2166.
Stukenbrock EH, and Mcdonald BA (2007) Geographical variation and positive diversifying
Syme RA, Tan K-C, Hane JK, Dodhia K, Stoll T, Hastie M, Furuki E, Ellwood SR, Williams AH, Tan Y-F, Testa AC, Gorman JJ, and Oliver RP (2016) Comprehensive annotation of the Parastagonospora nodorum reference genome using next-generation genomics, transcriptomics and proteogenomics. PLOS ONE 11:e0147221.
Syme RA, Tan K-C, Rybak K, Friesen TL, McDonald BA, Oliver RP, and Hane JK (2018) Pan-Parastagonospora Comparative Genome Analysis – effector prediction and genome evolution. Genome Biol Evol 10:2443–2457.
Talas F, Kalih R, Miedaner T, and McDonald BA (2016) Genome-wide association study identifies novel candidate genes for aggressiveness, deoxynivalenol production, and azole sensitivity in natural field populations of Fusarium graminearum. Mol Plant Microbe Interact 29:417–430.
Tilman D, Balzer C, Hill J, and Befort BL (2011) Global food demand and the sustainable intensification of agriculture. Proc Natl Acad Sci USA 108:20260–20264.
Torriani SFF, Melichar JPE, Mills C, Pain N, Sierotzki H, and Courbot M (2015) Zymoseptoria tritici: A major threat to wheat production, integrated approaches to control. Fungal Genet Biol 79:8–12.
United Nations (2019) Department of Economic and Social Affairs - Population Dynamics.
Vitti JJ, Grossman SR, and Sabeti PC (2013) Detecting natural selection in genomic data. Annu Rev Genet 47:97–120.
Williams JR, and Jones DG (1973) Infection of grasses by Septoria nodorum and S. tritici.
Transactions of the British Mycological Society 60:355–358.
Yang L, Zhu W, Wu E, Yang C, Thrall PH, Burdon JJ, Jin L, Shang L, and Zhan J (2016) Trade‐offs and evolution of thermal adaptation in the Irish potato famine pathogen Phytophthora infestans. Mol Ecol 25:4047–4058.
Zhan J, and McDonald B (2011) Thermal adaptation in the fungal pathogen Mycosphaerella graminicola. Mol Ecol 20:1689–701.
Zhan J, Stefanato FL, and McDonald BA (2006) Selection for increased cyproconazole tolerance in Mycosphaerella graminicola through local adaptation and in response to host resistance. Mol Plant Pathol 7:259–268.
Zhong Z, Marcel TC, Hartmann FE, Ma X, Plissonneau C, Zala M, Ducasse A, Confais J, Compain J, Lapalu N, Amselem J, McDonald BA, Croll D, and Palma-Guerrero J (2017) A small secreted protein in Zymoseptoria tritici is responsible for avirulence on wheat cultivars carrying the Stb6 resistance gene. New Phytol 214:619–631.
CHAPTER 1: Mutations in the CYP51 gene reduce DMI sensitivity in Parastagonospora nodorum populations in Europe and China
Danilo AS Pereira, Bruce A McDonald and Patrick C Brunner
Published as:
Pereira DA, McDonald BA, and Brunner PC (2017) Mutations in the CYP51 gene reduce DMI sensitivity in Parastagonospora nodorum populations in Europe and China. Pest Management Science 73:1503–1510. DOI: https://doi.org/10.1002/ps.4486
Abstract
Sterol demethylation inhibitors or DMIs have been widely used to manage agronomically important fungal diseases in wheat, but reports of DMI-resistant pathogens continue to mount.
Parastagonospora nodorum shows a wide range of sensitivity to DMIs, but until now no molecular mechanisms were identified to explain these differences. The aim of this study was to correlate the DMI sensitivity of a global collection of P. nodorum isolates with mutations in the CYP51 gene that encodes the target of DMI fungicides. Two non-synonymous mutations connected to DMI resistance in other plant pathogenic fungi were detected for the first time in the CYP51 gene of P. nodorum. The two mutations occurred at amino acid position 144, which is homologous to position 137 in other pathogens. The Y144F mutation was detected in China, Denmark, Sweden and Switzerland while the Y144H mutation was found in China and Switzerland. Both mutations were correlated with significantly reduced susceptibility to the DMI fungicide propiconazole. CYP51 mutations conferred reduced sensitivity against DMIs in field populations of P. nodorum originating from China, Denmark, Sweden and Switzerland.
Introduction
Sterol demethylation inhibitors, or DMIs, are fungicides that affect the biosynthesis of ergosterol in fungi (Siegel, 1981) and are extensively used in medicine and agriculture. DMIs inhibit a specific cellular pathway by binding to the sterol 14alpha-demethylase enzyme CYP51 (Délye et al., 1997). As a result of repetitive and extensive applications over time, the DMIs imposed strong selective pressure favoring the emergence of resistance in exposed pathogen populations (Zhan et al. 2006; Brunner et al. 2015). Resistance against DMIs is known to emerge through three main mechanisms (Cools et al., 2013): (i) CYP51 over- expression that increases the amount of the enzyme in the cell, (ii) increased activity of efflux pumps, often caused by over-expression of genes encoding membrane transporters that decrease the concentration of DMIs in fungal cells, and (iii) non-synonymous mutations in the CYP51 gene causing amino acid substitutions or deletions that affect the binding of the DMI to the enzyme. The combined occurrence of all three mechanisms has been reported in some strains of Candida albicans, a human pathogen (Morschhäuser, 2002). Mutations in the CYP51 gene are the most common mechanism reducing DMI sensitivity among plant pathogens (Cools and Fraaije, 2013). In field populations of the wheat pathogen Zymoseptoria tritici, causal agent of Septoria tritici leaf blotch, insertions and deletions in the promoter region, and more than 30 non-synonymous mutations in CYP51 gene have been correlated with reduced sensitivity to triazoles, a widely sprayed group of DMI fungicides (Cools and Fraaije, 2013).
Parastagonospora nodorum (synonyms: Phaeosphaeria nodorum, Stagonospora nodorum, Septoria nodorum) (Berk.) Quaedvlieg, Verkley & Crous, is the causal agent of Stagonospora or Septoria nodorum leaf and glume blotch (SNB) on durum and bread wheat (Solomon et al., 2006). The disease is found globally and field populations have been
al., 1997; Stukenbrock, Banke, and McDonald, 2006). While SNB is an important disease in some wheat production areas, a precise calculation of yield losses caused by SNB is controversial or absent in many countries (Oliver et al., 2012). The highest incidence of SNB is observed in Australia where losses in grain yield of winter wheat can reach up to 30%
(Bhathal et al., 2003). Due to low levels of resistance in wheat cultivars, fungicides are widely used to manage SNB (Solomon et al., 2006).
Although P. nodorum populations have been treated with many different groups of fungicides, including DMIs, reports of resistance are rare (Oliver et al., 2012). In an early study, Canadian populations of P. nodorum sampled in 1989 and 1990 were assessed (Peever et al., 2006). Some fields were treated with more than twice the recommended dose of propiconazole to increase selection pressure, but no resistant phenotypes of P. nodorum were found. Fifteen years later, Blixt et al. (2009) reported the sensitivity profiles of Swedish P.
nodorum populations sampled during 2003, 2004 and 2005. While Swedish isolates differed in sensitivity to propiconazole and other fungicides, the study could not establish a correlation between EC50 values and nucleotide substitutions in CYP51 and no alternative mechanisms were identified to explain differences in DMI sensitivity among isolates.
The study by Blixt et al. (2009) included only partial sequences of the CYP51 gene and considered only Swedish isolates. Here, we extended their approach by sequencing the entire CYP51 gene, including flanking regions, and by assessing a large number of P. nodorum isolates sampled from wheat fields around the world.
Experimental methods
Origins of P. nodorum isolates and DNA extraction
All populations of P. nodorum used in this study were collected from naturally infected wheat fields. Locations and collection dates are summarized in Table 1. Isolates were retrieved from long-term storage on silica gel and grown on yeast malt sucrose agar (YMA, 4 g L-1 yeast extract, 4 g L-1 malt extract, 4 g L-1 sucrose, 12 g L-1 agar and 50 mg L-1 kanamycin). Isolates were grown on Petri dishes for approximately 7 days at 18°C in the dark. For DNA extraction, fungal mycelium was transferred to Erlenmeyer flasks containing 25 ml of yeast sucrose broth (YSB; 10 g L-1 yeast extract and 10g L -1 sucrose) and grown on a shaker for 4 days at 120 rpm at 18°C in the dark. Fungal tissue was lyophilized, ground into a powder and total DNA was extracted using the DNeasy Plant Mini DNA extraction kit (Qiagen GmbH, Hilden, Germany) following the manufacturer’s instruction.
CYP51 gene amplification and sequencing
Based on genomic sequences from the P. nodorum reference isolate SN15 v2.0 (Hane et al., 2007) four pairs of primers with overlapping target regions were designed for the CYP51 gene with the Primer3 software (Untergasser et al., 2012). The primers spanned the entire coding region of the gene including approximately 430 base pairs of the upstream flanking region (Supplementary Figure 1; Supplementary Table 1; http://genome.jgi.doe.gov/cgi- bin/dispTranscript?db=Stano2&table=protein&id=10188&useCoords=1&width=70&padding
=500). The full sequence was achieved with eight sequencing reactions for each isolate.
PCR amplification was performed in a total volume of 25 µL. The PCR mix contained 0.5 µM of each forward and reverse primer (Supplementary Table 1), 0.25 ng DNA µL-1, 0.25 µM of dNTPs, 2 µL of 10X PCR buffer (Fermentas) and 0.1 U µL-1 of Dream Taq DNA Polymerase (Fermentas). The PCR cycle parameters were: 2 min of initial denaturation at 96°C followed by 35 cycles of 96°C for 30 s, annealing at 56°C for 30 s, 72°C for 1 min and final
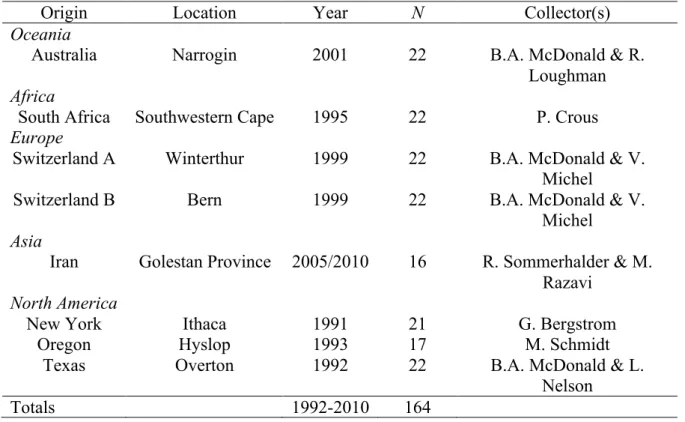
Table 1 Origin, year of collection and sample size (N) of Parastagonospora nodorum populations included in analyses of CYP51 sequences.
Origin Abbreviation Year Location N Collectors
Asia
China CN 2001 Fujian Province 16 R. Wu
Iran IR 2005/2010 Golestan Province 19 R. Sommerhalder & M. Razavi Europe
Denmark DK 2005 Jylland 34 E. Stukenbrock
Sweden SE 2005 Uppsala 44 E. Blixt
Switzerland CH94 1994 Winterthur 71 S. Keller
CH99 1999 Winterthur 53 B.A. McDonald & V. Michel North America
New York NY 1991 Ithaca 20 G. Bergstrom
Oregon OR 1993 Hyslop 10 M. Schmidt
Texas TX 1992 Overton 20 B.A. McDonald & L. Nelson
Oceania
Australia AU 2001 Narrogin 15 B.A. McDonald & R. Loughman Africa
South Africa SA 1995 Southwestern Cape 19 P. Crous
Total / Average 1992-2010 321
remove unincorporated nucleotides and primers using NucleoFast 96 PCR plates (Macherey- Nagel, Oensingen, Switzerland).
Sequencing reactions were conducted in a 10 µL volume using BigDye Terminator v3.1 Sequencing Standard Kit (Life Technologies, Applied Biosystems, Grand Island, NY, USA) with both respective forward and reverse primers. The cycling parameters were 96°C for 2 min followed by 99 cycles of 96°C for 10 s, 50°C for 5 s and 60°C for 4 min. PCR products were cleaned with the illustra Sephadex G-50 fine DNA Grade column (GE Healthcare, Pittsburgh, PA, USA) according to the manufacturer’s recommendations and sequenced with an ABI 3130xl Genetic Analyzer (Life Technologies, Applied Biosystems). Assembly of forward and reverse reads and alignment of sequences was performed using the program package Geneious (Biomatters, Auckland, New Zealand) and the implemented MUSCLE algorithm.
The phylogenetic reconstruction was inferred by using the Maximum Likelihood method based on the General Time Reversible model. The unrooted tree with the highest log likelihood (-3364.1056) is shown. Evolutionary analyses were conducted in MEGA7 (Kumar et al., 2016).
Fungicide assay
First, an exploratory phenotyping experiment was performed with two isolates each from the CN (China), IR (Iran), CH99 (Switzerland), NY (New York, USA), TX (Texas, USA), AU (Australia) and SA (South Africa) populations and five doses of propiconazole (Syngenta, Basel, Switzerland) diluted in DMSO (Supplementary Figure 2). For this assay, 5-mm diameter plugs of colonized YMA were transferred from 7-days-old colonies to YMA plates amended either with fungicide or only DMSO. The propiconazole doses were 0.1, 0.5, 1, 5 and 10 mg litre-1. After inoculation, plates were randomized in a growth chamber set to 18°C with 70%
inoculation (DPI) to determine the optimal time points for digital image analysis. Due to complete inhibition of mycelial growth at the two highest doses (Supplementary Figure 2), propiconazole concentrations of 0.1, 0.5 and 1 mg litre-1 were selected for the subsequent main experiment. Selected time points for digital image analysis were 3, 6 and 9 DPI.
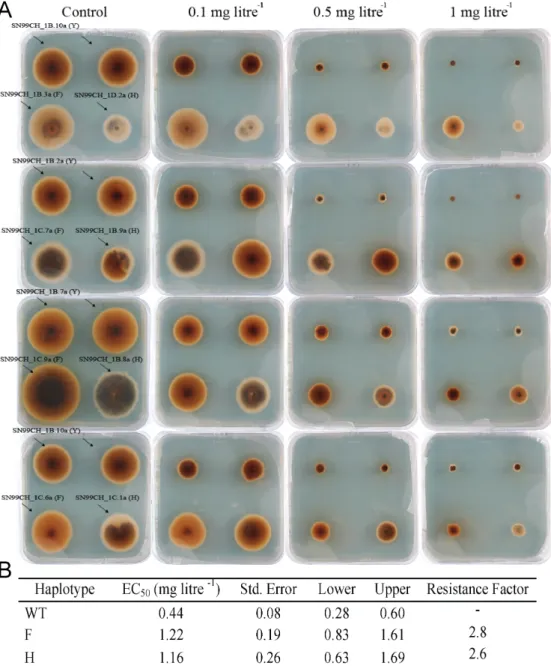
In the main experiment, growth rates of mutant and wild-type isolates were compared to measure the fitness advantage or cost of carrying the mutation at different fungicide concentrations. The experimental design is shown in Figure 1. The experiment was conducted on square Petri plates using three isolates possessing the Y144 haplotype, four isolates possessing the Y144H haplotype and four isolates possessing the Y144F haplotype. The 11 Swiss isolates were sampled from the same randomly mating population, with the CYP51 variants present in different genetic backgrounds that were selected in the same field environment. Mycelial plugs were placed on square Petri plates containing potato dextrose agar (PDA, 4g L-1 potato starch, 20 g L-1 dextrose, 15 gL-1 agar and 50 mg L-1 kanamycin) amended either with propiconazole or pure DMSO. Each square plate contained two fungal plugs of a wild-type isolate (Y144), one of a haplotype H isolate (Y144H) and one from a haplotype F isolate (Y144F). Each experiment was repeated four times.
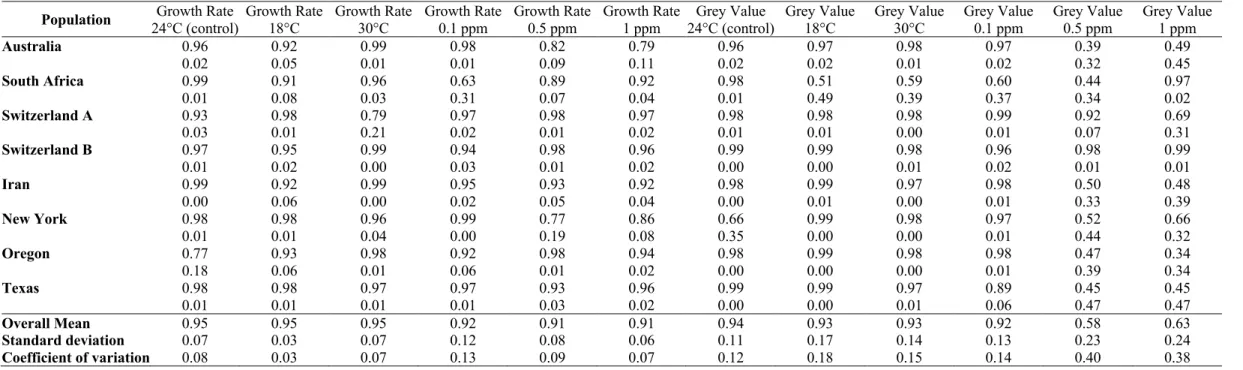
Images used for phenotyping were captured through the Petri dish lid using standardized camera settings and lighting environments described earlier (Lendenmann et al., 2014). After image capture, Petri dishes were re-randomized and returned to the growth chamber for further incubation until the next image acquisition. Digital images were processed using a batch macro developed in the open-source software ImageJ (Schneider et al., 2012). The average colony radius for each isolate and colony age was calculated by dividing the average colony area by π and taking the square root. The radial growth rate (mm
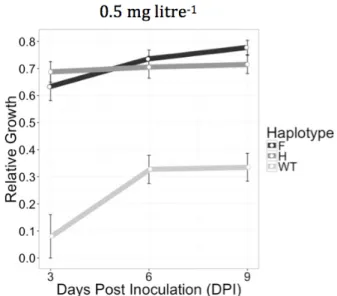
Figure 1. (A) Parastagonospora nodorum growth in the absence and presence of different doses of propiconazole. The name of the isolates is followed by the amino acid present in the 144 position, wild type (Y) and mutants (F and H). Each Petri dish in the same row contains the same three isolates. Each Petri dish in the same column contains different isolates, except for the wild types where only three isolates were available. This experiment was repeated four times. Colonies were photographed at 9 days post inoculation. (B) EC50 values (mg litre-1) of propiconazole and resistance factor for the three haplotypes used in the main experiment.
day -1) for each isolate was measured by plotting the colony radius over time and applying a general linear model using Pearson’s correlation coefficient in R (R Core Team, 2019). The slope of the regression line is the radial growth rate (Supplementary Figure 3). Fitness costs or advantages associated with CYP51 mutations were approximated as the ratios of radial growth rates on fungicide-amended media to fungicide-free controls. The EC50 values were determined based on the mycelial growth of the 11 isolates used in the main experiment at 9 DPI using the package ‘DRC’ in R and the resistance factors (RF) were determined by dividing the EC50 of the resistant haplotype by the EC50 of the wild-type (Ritz et al., 2015).
Results
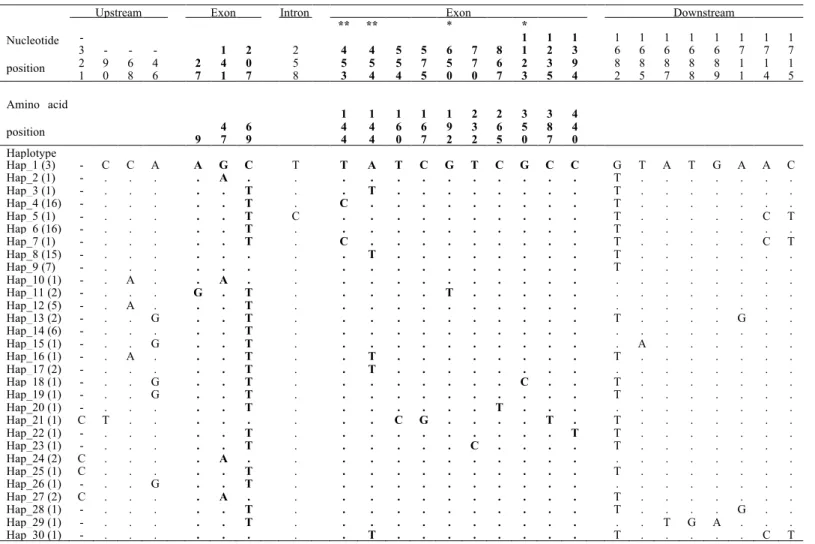
The CYP51 gene was analyzed in 321 P. nodorum isolates. The complete CYP51 gene (1673 bp) including 5’ upstream (401 bp) and 3’ downstream regions (42 bp) were sequenced for 96 isolates. An additional 225 isolates were partially sequenced for a 723 bp region covering the variable sites associated with DMI resistance. A total of 26 sites (1.2%) were polymorphic, resulting in 30 multilocus haplotypes for the entire gene (Table 2). Two non-synonymous substitutions were detected at codon 144. One nucleotide substitution changed the wild-type codon from TAC to TTC and the corresponding amino acid from tyrosine to phenylalanine (Y144F). The other substitution changed TAC to CAC and the amino acid to histidine (Y144H).
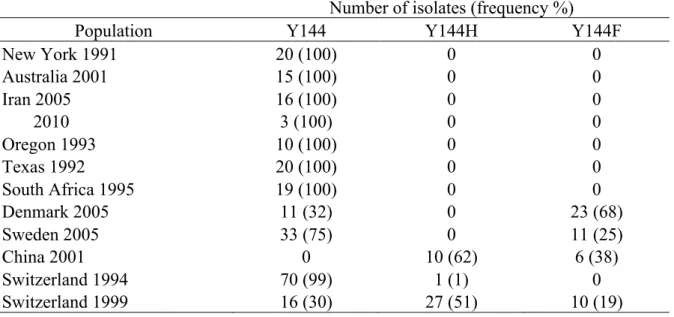
The Y144F mutation was found in isolates from Switzerland, Sweden, Denmark and China. The Y144H mutation was restricted to Switzerland and China (Table 3). A significant increase in frequency of isolates carrying the two mutations was observed in Switzerland between 1994 and 1999. The Y144H mutation increased in frequency from 1% in the 1994 collection to 51% in the 1999 collection. Similarly, the Y144F mutation increased from 0%