Synthese, Struktur und spektroskopische Eigenschaften selen- und tellurhaltiger topologischer Isolatoren und thermoelektrischer Materialien
195
0
0
Volltext
(2)
(3)
(4)(5)
(6)
(7)
(8)(9)
(10)
(11)
(12)(13)
(15)
(16)
(17)
(18)(19)
(20)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(33)
(35)
Abbildung
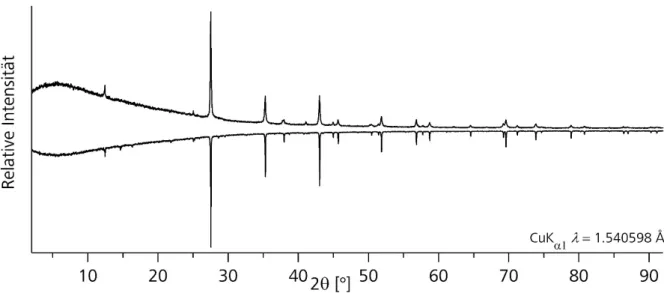
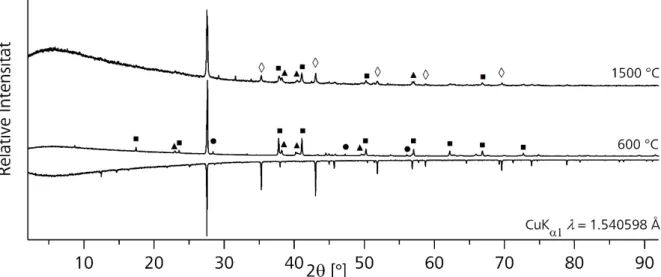
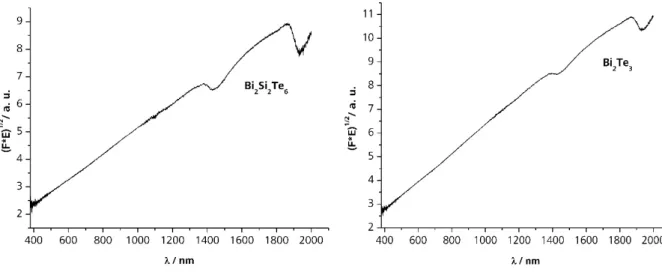
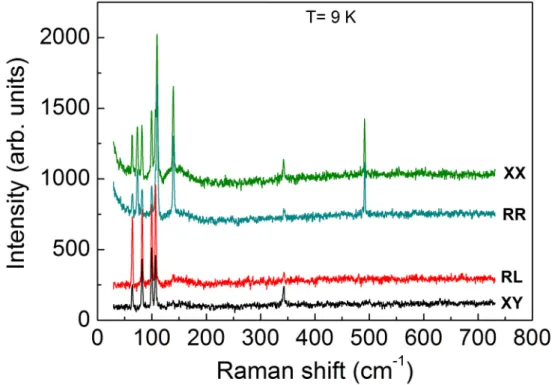
+7
ÄHNLICHE DOKUMENTE
Im Rahmen dieser Arbeit soll die Möglichkeit untersucht werden, durch optische Anregung von Elektronen oder durch Beschleunigung der Elektronen in elektrischen Feldern einen
Im Vergleich zu 1-Octen (Abb. 3.1) wird deutlich, dass 1-Octen noch besser in das Terpolymer eingebaut wird, was aber damit verbunden ist, dass ein Teil des 1-Butens bei der
The strong changes between the PM and AFM states calculated and observed for EuRh 2 Si 2 indi- cate that the formation of the AFM state in YbRh 2 Si 2 very likely also results in
Die Elektroaffinität des Bi'"—Ions ist wenig ausgesprochen; es läßt sich leicht zu Metall reduzieren, namentlich durch die alkalischen Lösungen von Zinnhydroxydul.. 3 Sn02” +
1 Allerdings wurden diese topologischen Zustände in Materialien gefunden, in denen Wechselwirkungen zwischen den Elek- tronen vernachlässigt werden können.. Wie be- reits
Die Synthese der Liganden 3a und 3b wird im Rahmen dieser Arbeit nachvollzogen, da diese, wie im folgenden Kapitel beschrieben werden wird, zur Komplexsynthese verwendet
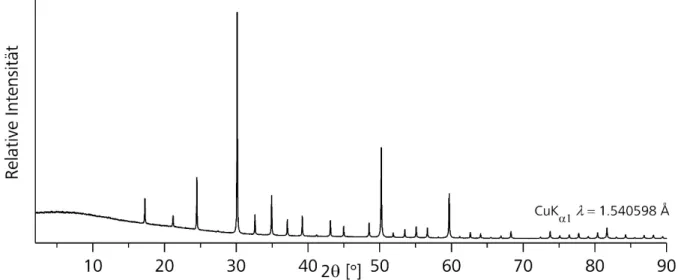
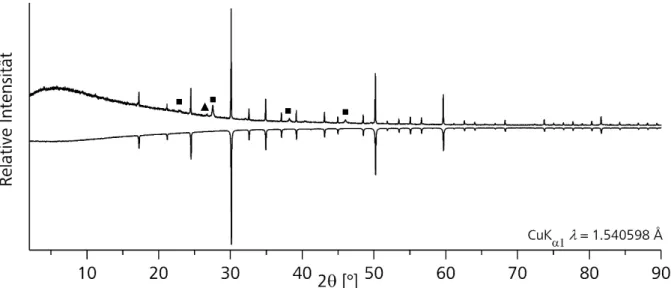
Diese Methode unterscheidet sich von Methode 4 dadurch, dass nach dem ersten Reaktionsschritt die Ampulle nicht abgekühlt wurde.. Stattdessen wurde das Chalkogen portionsweise zu
Definition: Alle Atome eines Liganden werden nach ihrer topologischen Entfernung Sphären zugeordnet.. Alle Atome, die in
