M ASTERARBEIT
Technische Universität Dresden
Fakultät Mathematik und Naturwissenschaften Fachrichtung Chemie und Lebensmittelchemie
Professur für Radiochemie
eingereicht von Luisa Köhler geb. am 27.01.1995 in Jena
Betreuer/Gutachter Prof. Dr. Thorsten Stumpf Betreuer/Gutachter Dr. Juliane März
Dresden, den 19.10.2018
ATR-FT-IR-Spektroskopie engl. Attenuated Total Reflection Fourier Transform Infrared Spectroscopy, abgeschwächte Totalreflexion Fourier Transform Infrarot Spektroskopie
äq Äquivalente
Dipp2Im 1,3-Bis(2,6-diisopropylphenyl)imidazol-2-
yliden
HSQC engl. Heteronuclear Single Quantum
Corherence, heteronuklear korreliertes NMR-Experiment
iPr Isopropylgruppe
iPr2Im 1,3-Diisopropylimidazol-2-yliden
IR Infrarot
m engl. middle, mittel
MeCN Acetonitril
NMR engl Nuclear Magnetic Resonance
Spectroscopy, Kernresonanzspektroskopie
s Singulett-Zustand
s engl. strong, stark
SC-XRD engl. Single Crystal X-Ray Diffractometry,
Einkristallröntgendiffraktometrie
t Triplett-Zustand
THF Tetrahydrofuran
TMS Tetramethylsilan
ÜM Übergangsmetall
vs engl. very strong, sehr stark
vw engl. very weak, sehr schwach
w engl. weak, schwach
II I NHALTSVERZEICHNIS
1 EINLEITUNG ... 1
2 STAND DER FORSCHUNG ... 3
3 AUSWERTUNG UND DISKUSSION ... 8
3.1 Theoretische Betrachtungen ... 8
Carbenstabilität ... 8
Strukturoptimierung ... 10
3.2 Charakterisierung von Uran-Komplexen mit 1,3-Diisopropylimidazol-2-yliden .... 11
Einkristallröntgendiffraktometrie ... 11
Infrarot-Spektroskopie ... 18
NMR-Spektroskopie ... 22
3.3 Charakterisierung von Uran-Komplexen mit 1,3-Bis(2,6- diisopropylphenyl)imidazol-2-yliden ... 24
Einkristallröntgendiffraktometrie ... 24
IR-Spektroskopie ... 29
NMR-Spektroskopie ... 31
3.4 Vergleichende Studien mit Übergangsmetallen und 1,3-Diisopropylimidazol-2- yliden 35 Einkristallröntgendiffraktometrie ... 35
IR-Spektroskopie ... 35
NMR-Spektroskopie ... 36
3.5 Vergleichende Studien mit Übergangsmetallen und 1,3-Bis(2,6- diisopropylphenyl)imidazol-2-yliden ... 37
Einkristallröntgendiffraktometrie ... 37
IR-Spektroskopie ... 41
NMR-Spektroskopie ... 42
4 ZUSAMMENFASSUNG UND AUSBLICK ... 45
5 EXPERIMENTELLER TEIL ... 47
5.1 Allgemeines Synthesevorgehen ... 47
5.2 Synthesen der An(IV)-Komplexe ... 48
5.3 Synthesen der Übergangsmetallkomplexe ... 50
5.4 Kristallzucht ... 53
5.5 Geräte, Methoden und Chemikalien ... 54
NMR-Spektroskopie ... 54
Infrarot-Spektroskopie ... 54
Einkristallröntgendiffraktometrie ... 54
III LITERATURVERZEICHNIS ... 56
IV DANKSAGUNG ... 58
V ANHANG ... 59
Theoretische Betrachtungen ... 59
IR-Spektroskopie ... 59
Kristallstrukturbestimmung ... 64
SELBSTSTÄNDIGKEITSERKLÄRUNG ... 66
1
1 E INLEITUNG
Bei der friedlichen Nutzung von Kernenergie zur Elektrizitätsgewinnung fallen Abfallprodukte an, die signifikante Konzentrationen an langlebigen, radioaktiven Elementen enthalten. Diese sind bspw. die Actinide U, Pu, Am und Np.[1] Die Suche nach einem geeigneten Endlager für diesen Abfall ist unabdingbar, um das durch sie bestehende Gefahrenpotenzial zu minimieren.
Grund dafür ist die emittierte ionisierende Strahlung, die Schäden an Umwelt und dem menschlichen Organismus hervorrufen kann. Zu diesem Zeitpunkt ist die Frage über einen geeigneten Standort für ein solches Endlager jedoch noch nicht abschließend geklärt. Während der Langzeitlagerung können die Actinide unterschiedlichen Prozessen ausgesetzt sein, wie der Präzipitation, Sorption und Kolloidbildung, sowie Komplexierung.[2] Vor allem die Koordination zu organischen bzw. anorganischen Liganden beeinflusst die Mobilität und damit die Migration der Actinide massiv.[3] Diese ist ein großes Problem, da es so zu einer Freisetzung und Distribution der radioaktiven Bestandteile in die Umwelt kommen könnte, wodurch Ressourcen weitreichend kontaminiert würden. Durch fundiertes Wissen über die Chemie der Actinide können diese Prozesse verstanden und möglicherweise sogar vorhergesagt werden. Da in der reduktiven Umgebung eines möglichen Endlagers mehrere hundert Meter unter der Erdoberfläche die Bildung vierwertiger Actinide bevorzugt ist, steht deren chemisches Verhalten im Kontext der Koordination im Fokus.[4] Um ein Grundverständnis für die Koordinationschemie dieser Spezies zu erlangen, ist es notwendig die Bindungssituationen zwischen Actiniden und harten bzw. weichen organischen oder anorganischen Liganden zu untersuchen und zu interpretieren. Um die komplexen Prozesse verstehen zu können, bietet es sich an, zunächst ein kleines und einfach zu untersuchendes System zu betrachten. Darin kann der Fokus getrennt auf bestimmte, einzelne Effekte gelegt werden, die in natürlichen Systemen gemeinsam auftreten könnten. Dies geschieht durch die Etablierung kleiner und leicht zu untersuchender Modellliganden, woraufhin anschließend die gewonnenen Erkenntnisse auf große, natürliche Systeme übertragen werden könnten. Das Bindungsmotiv zu weichen Donorliganden ist dabei besonders wenig untersucht, sodass dieses im Fokus dieser Arbeit steht.
Ziel dieser Arbeit ist die Eruierung des Systems von vierwertigem Uran mit weichen Donorliganden im Vergleich zu analogen Komplexen mit den tetravalenten Übergangsmetallen Zr und Hf. Als weiche Donorliganden werden hierbei die N-heterocyclischen Carbene 1,3- Diisopropylimidazol-2-yliden (iPr2Im) und 1,3-Bis(2,6-diisopropylphenyl)imidazol-2-yliden (Dipp2Im) verwendet, da diese ausschließlich weiche σ-Donoreigenschaften aufweisen.[5]
Neben der Synthese der freien Carbene sollen vor allem die entsprechenden Metallkomplexe dargestellt werden. Anschließend soll die Charakterisierung in Lösung via NMR und im Festkörper via SC-XRD und IR-Spektroskopie erfolgen. Die verwendeten Methoden sollen dann bezüglich ihrer Eignung eingeschätzt werden. Die experimentellen Arbeiten sollen zudem
2
3
2 S TAND DER F ORSCHUNG
Die Substanzklasse der Carbene zeichnet sich durch ein divalentes, neutrales Kohlenstoffatom aus, welches nur sechs Valenzelektronen besitzt.[5] Die Hybridisierung am Carben-Zentrum entscheidet darüber, ob eine lineare oder gewinkelte Umgebung zu finden ist. Der Großteil der Carbene ist am Kohlenstoffatom sp2-hybridisiert und liegt in einer gewinkelten Form vor. Bei einer linearen Geometrie liegt hingegen ist eine sp-Hybridisierung zu finden, wobei dieser Fall jedoch sehr selten anzutreffen ist. Der Grund dafür ist, dass das durch die sp2-Hybridisierung gebildete σ-Orbital partiellen s-Charakter erhält und damit relativ zum ehemals vorhandenen p- Orbital energetisch erniedrigt wird. Die beiden ungepaarten Elektronen am Carben-Kohlenstoff können das leere σ-Orbitale mit parallelem Spin besetzen, woraufhin ein Singulett- Grundzustand (σ1pπ1, 3B1) entsteht (Abbildung 1 1). Werden jedoch beide leere Orbitale in antiparallelem Spin besetzt, resultiert ein Triplett-Grundzustand (σ2pπ0, 1A1) (Abbildung 1 3).[5]
Abbildung 1 Mögliche Elektronenkonfigurationen am Carben-Kohlenstoff und Stabilisierung von Singulett- Carbenen.[6]
Die Eigenschaften und Reaktivität der Carbene wird durch die Multiplizität des Grundzustandes bestimmt, der seinerseits durch die Energien der σ- und pπ-Orbitale determiniert wird.[7] Bei einem Energieunterschied zwischen dem σ- und dem pπ-Orbital von 2 eV wird ein Singulett- Grundzustand erreicht, während ein Energieunterschied kleiner als 1,5 eV zum Triplett- Grundzustand führt.[8] Donorsubstituenten können Singulett-Carbene stabilisieren, da sie die Elektronenlücke im leeren pπ-Orbital teilweise auffüllen können (Abbildung 1 2).[6] Daraus resultiert nucleophile Reaktivität, während Triplett-Carbene aufgrund ihrer ungepaarten Elektronen als Diradikale bezeichnet werden können.[5,9] Als besonders effektive Donorgruppen zur Singulett-Stabilisierung haben sich Aminosubstituenten am Carben- Kohlenstoff erwiesen.[10] Durch Wechselwirkungen der π-Elektronen der Substituenten mit dem pπ-Orbital am Carben-Zentrum kommt es zur Ausbildung eines vier-Elektronen-drei- Zentren-π-Systems. Die N–C-Bindungen weisen dabei teilweisen Mehrfachbindungscharakter auf.[5] Exemplarisch dafür lassen sich in N-heterocyclischen Verbindungen Aminosubstituenten am Carben-Kohlenstoff finden. Bekannte Vertreter dieser Verbindungen sind die N- heterocyclischen Fünfringcarbene. Die Stickstoffatome im Heterocyclus sind für die elektronische Stabilisierung des Carben-Zentrums ausreichend, sodass eine weitere Stabilisierung durch sterisch anspruchsvolle Substituenten nicht nötig ist.
4 einen Singulett-Grundzustand ausbilden.[5] WANZLICK versuchte 1960 durch α-Eliminierung von Chloroform aus 4 ein NHC zu isolieren, gelangte dabei jedoch nur zu dem Dimer 5 (Abbildung 2).[11] Das gebildete Carben war demnach zu reaktiv, als dass es isoliert werden konnte.
Abbildung 2 Von WANZLICK durchgeführte α-Eliminierung aus 4.[11]
Eine weitere Stabilisierung wurde in der Existenz eines delokalisierten π-Systems vermutet, wie es in ungesättigten Analoga zu finden ist. Die Wiederholung des Experiments mit 6 führte allerdings ebenfalls nicht direkt zum Erfolg. Das freie Carben konnte zwar nicht isoliert, jedoch mit Hilfe von Quecksilber(II)acetat stabilisiert werden, sodass sich der entsprechende Metallkomplex 7 bildete (Abbildung 3).
Abbildung 3 Stabilisierung von ungesättigten Imidazol-2-ylidenen mit Quecksilber.[12]
Letztlich gelang es ARDUENGO 1991, das erste stabile N-heterocyclische Singulett-Carben, das 1,3-Di-1-adamantylimidazol-2-yliden, zu isolieren und zu kristallisieren.[13]
Imidazol-2-ylidene 9 können allgemein auf zwei unterschiedlichen Wegen synthetisiert werden (Abbildung 4). Zum einen durch Deprotonierung der entsprechenden Imidazoliumsalze 8, zum anderen durch reduktive Entschwefelung analoger Thione 10.[5,13]
4 5
6 7
5
Abbildung 4 Synthese von Imidazol-2-ylidenen durch Deprotonierung der Imidazoliumsalze oder durch reduktive Entschwefelung.[6,14]
Als Base wurde von ARDUENGO zunächst Natriumhydrid mit katalytischen Mengen Dimethylsulfoxid eingesetzt.[13] Andere Basen, wie Kaliumhydrid, Lithiumdiisopropylamid oder Lithiumtetramethylpiperidin sind ebenfalls einsetzbar.[6] Diese Isolierungsmöglichkeiten zeigen, dass es sich bei Carbenen nicht nur um reaktive Intermediate handelt, sondern dass sie gezielt in bspw. Komplexierungsreaktionen eingesetzt werden können.
Die Imidazol-2-ylidene bilden die wohl größte Gruppe der NHCs. Nicht nur das erste isolierte Carben gehört zu dieser Substanzklasse, sondern auch die in der vorliegenden Arbeit betrachteten Carbene. Es handelt sich dabei um das 1,3-Diisopropylimidazol-2-yliden (iPr2Im, 9a) und das 1,3-Bis(2,6-diisopropylphenyl)imidazol-2-yliden (Dipp2Im, 9b).
Abbildung 5 In der Arbeit verwendete NHCs. Links: 1,3-Diisopropyliimidazol-2-yliden, rechts: 1,3-Bis(2,6- diisopropylphenyl)imidazol-2-yliden.
Eine ähnliche Struktur, die allerdings kleinere Methylsubstituenten besitzt, ist bereits synthetisiert und charakterisiert worden.[15] Typischerweise lässt sich ein N–C–N Bindungswinkel von 101,5(2)-101,2(2)° und ein verhältnismäßig großer C–N Abstand von 136 pm finden.[6,15] Die freien Carbene weisen im Vergleich zu den Imidazoliumionen eine um ca.
5 pm verlängerte C–N Bindung auf, was auf eine Schwächung der π-Delokalisierung durch die Deprotonierung hinweist.[5] Dies verdeutlicht noch einmal, dass hauptsächlich die thermodynamische (elektronische Stabilisierung durch die Stickstoffatome) und nicht die kinetische Stabilisierung (sterische Hinderung) die Isolierung von Carbenen ermöglicht.[5]
8 9 10
iPr2Im Dipp2Im
9a 9b
6 werden. Fischer-Carben Komplexe weisen in der Regel folgende Merkmale auf: heteroatomare Stabilisierung am Carben-Zentrum; ein gebundenes Metall in niedriger Oxidationsstufe; π- Akzeptorliganden und elektrophilen Charakter des Carben-Kohlenstoffes. Im Gegensatz dazu zeichnen sich Schrock-Carbene durch Metalle in höheren Oxidationsstufen, Alkylgruppen oder Wasserstoffatome und nukleophilen Charakter am Carben-Zentrum aus. Bindungen in Schrock-Carbenen können als kovalente Doppelbindung zweier Triplett-Zentren verstanden werden.[16,17] NHCs bilden ihre eigene Carben-Gruppe, zeigen jedoch Merkmale, die dazu führen, dass sie als Fischer-Carben Analoga eingestuft werden können. Anlass dazu gibt bspw.
die Tatsache, dass das Carben-Zentrum durch die Benachbarung der Stickstoffatome im Heterocyclus stabilisiert wird. Des Weiteren weisen NHCs σ-Donor Eigenschaften auf und bilden allgemein dative Donor-Akzeptor Wechselwirkungen mit dem Metall aus. Ob jedoch auch eine für Fischer-Carbene charakteristischen π-Rückbindung vorliegt, ist noch nicht abschließend geklärt.[17]
Sicher ist in jedem Falle, dass NHCs als weiche Liganden für Übergangsmetalle, Lanthanide und Actinide eingesetzt werden können.[14,17–20] Prominente Beispiele sind als Katalysatoren eingesetzte Übergangsmetallkomplexe. Exemplarisch hierfür ist der für die Olefinmetathese entwickelte Ruthenium-Carben Komplex zu nennen, für den 2005 der Nobelpreis verliehen wurde.[21–23]
Der erste U(IV)-NHC Komplex (11) wurde erst 2004 von EVANS publiziert (Abbildung 6). In diesem Komplex finden sich neben einem Tetramethylimidazol-2-yliden zwei Cp*
(Pentamethylcyclopentadien)-Liganden und ein doppelt gebundenes Sauerstoffatom. Die U–
CCarben Bindungslänge beträgt 2,636(9) Å.[24]
Abbildung 6 Erster dokumentierter NHC Komplex (C5Me5)2U(O)[C(NMeCMe)2].[24]
Zwar gibt es bis zum jetzigen Zeitpunkt noch keinen U(IV)-Komplex mit 9a als Ligand, es sind allerdings bereits Übergangsmetallkomplexe bekannt. NIEHUES et al. setzte tetravalentes Titan, Zirkonium und Hafnium jeweils als Übergangsmetallchlorid-THF-Addukt mit dem zuvor deprotonierten Imidazoliumsalz um. Dabei gelangte er zu den ÜM-Komplexen, die oktaedrisch
11
7 von vier Chloro- und zwei Carben-Liganden umgeben sind. Die Chloroliganden befinden sich dabei in einer Ebene. Es lassen sich M–CCarben Abstände von ca. 2,4 Å und C–M–C Bindungswinkel von nahezu 180° finden.[25]
Abbildung 7 Darstellung des [Zr(iPr2Im)2Cl4] Komplexes.[25]
Bezüglich des Dipp2Im Carbens ist 2009 von GARDNER der entsprechende U(IV) Komplex beschrieben worden (Abbildung 8, 13). Das entsprechende Carben kann käuflich erworben werden und wurde mit UCl4 in THF umgesetzt. Daraus resultierten hellgrüne Kristalle. Das Uran-Zentrum ist dabei ähnlich wie in 12 von vier Chloroliganden in einer Ebene und zwei Carbenen in oktaedrischer Koordinationssphäre umgeben. Die Carbene sind dabei im Vergleich zu 12 nicht in die gleiche Richtung ausgerichtet, sondern um 78,5° gegeneinander verdreht.[26]
Als Grund dafür kann die Vermeidung von repulsiven Wechselwirkungen der Isopropylgruppen am Phenylsubstituenten angesehen werden.
Abbildung 8 Von GARDNER et al. publizierter [U(Dipp2Im)2Cl4]∙THF Komplex.[26]
12
13
8
3.1 Theoretische Betrachtungen
Carbenstabilität
Da die Erzeugung des Carbens für die Bildung des Metall-Komplexes von großer Bedeutung ist, spielt die Stabilität der freien Carbene ebenfalls eine wichtige Rolle. Um die Stabilität eines Carbens zu beurteilen, können drei Parameter zur Hilfe genommen werden. Diese sind die Energielücke zwischen s (Singulett-Zustand) und t (Triplett-Zustand) (Es-t), die Carben- Stabilisierungsenergie (CSE) und die Dimerisierungsenergie (Edim). Es-t ist dabei die Differenz der Energien der optimierten Singulett- und Triplettstrukturen.[27] Je kleiner diese Energie ist, desto weniger Energie ist für den Übergang vom Singulett- zum Triplettzustand nötig, was einen Einfluss auf die Reaktivität haben kann.
Die CSE wird als Reaktionsenergie der folgenden Reaktion definiert:
Abbildung 9 Modellreaktion zur Berechnung der Carben-Stabilisierungsenergie (CSE).[27]
Es wird dabei die Stabilität der Carbene im Vergleich zum kleinstmöglichen Carben, dem Methylen, betrachtet. Ein Carben ist umso stabiler, je größer die CSE ist.[27] Je größer die Reaktionsenergie ist, desto endothermer ist die Reaktion, sodass das Carben erhalten bleibt und damit nicht protoniert wird. Nicht ausreichend stabilisierte Carbene neigen zu einer Dimerisierung unter Ausbildung einer C–C-Doppelbindung, sodass ein elektronenreiches Olefin bzw. Entetaamin entsteht (Abbildung 10).[5,28]
Abbildung 10 Dimerisierungsreaktion von zwei Carbenen.
Besonders die gesättigten Vertreter fünfgliedriger NHCs (14) sind davon betroffen, während ihre ungesättigten Analoga (15) durch die 6π-Elektronen-Delokalisierung stabilisiert werden (Abbildung 11).[29]
R = iPr (9a) Dipp (9b) Me (9c)
9 9=9
9
Abbildung 11 Darstellung von gesättigten (links) und ungesättigten (rechts) NHCs.
Deshalb ist die Dimerisierungsenergie Edim ein Parameter, um die Stabilität von Carbenen zu beschreiben. Je größer die Werte der Edim ist, desto stabiler ist das Carben. Es gilt zu beachten, dass diese Parameter nur eine Aussage über die thermodynamische, aber nicht über die kinetische Stabilität zulassen.[27]
Es wird zunächst angenommen, dass es sich bei Dipp-substituierten Carben 9b um das stabilere Carben im Vergleich zum Isopropyl-substituierten Carben 9a handelt, da für dieses bereits Kristallstrukturdaten verfügbar sind und durch die sterisch sehr anspruchsvollen Dipp- Substituenten bspw. eine Dimerisierung nur sehr schwer ablaufen könnte. Zusätzlich zu den bereits vorgestellten Carbenen wird in die Betrachtungen das 1,3-Dimethylimidazol-2-yliden (9c) einbezogen, damit mit einem dritten Vertreter eventuelle Trends besser erkennbar und einschätzbar sind. Die mittels DFT ermittelten Werte sind in Tabelle 1 zusammengefasst.
Tabelle 1 Theoretisch Berechnete Werte der Carbenstabilitätsparameter.
*Diese Daten wurden mit einer anderen Methode berechnet (s. Anhang)
Bei Betrachtung der Es-t wird zunächst deutlich, dass die Carbene mit den kleineren Resten einen identischen Wert von 90 kcal∙mol−1 aufweisen. Durch die große Ähnlichkeit der Substituenten war dies auch zu erwarten. Im Vergleich dazu liegt der Wert beim Dipp2Im nur bei 50 kcal∙mol−1, was bedeutet, dass der Singulett-Zustand bei 9a und 9c besser stabilisiert ist, als in jenem mit den Dipp-Substituenten. Wird das Augenmerk auf Edim gelegt, so zeigen die methyl- und isopropylsubstituierten Carbene einen identischen Wert, der mit 10 kcal∙mol−1 wesentlich kleiner ist, als die 50 kcal∙mol−1 von 9b. Damit wird die Vermutung bestätigt, dass das Carben mit den sterisch anspruchsvolleren Diisopropylphenyl-Resten stabiler bezüglich der Dimerisierung ist als 9a und 9c. Besonders deutlich wird dies, wenn die theoretische Struktur für das Dipp2Im-Dimer 9b=9b betrachtet wird. Zwar werden die Phenylringe so gedreht, dass es zu π-π-Wechselwirkungen kommen kann, jedoch sind die repulsiven Interaktionen zu groß, als dass sich diese Struktur bei Raumtemperatur bilden könnte.
Für alle Carbene ist die Edim positiv, was bedeutet, dass die Dimerisierungsreaktion endotherm ist. Damit sollten alle betrachteten Vertreter als freie Carbene und nicht als Dimer vorliegen, wodurch sie für eine Komplexierung frei zur Verfügung stehen sollten. Die Werte für die CSE
Parameter
[kcal mol−1] Me2Im (9c) iPr2Im (9a) Dipp2Im (9b)
Es-t 90 90 50
Edim 20/10* -/10* -/50*
CSE 100 110 110
••
••
14 15
10 der Dimerisierung und befinden sich im Singulett-Zustand.
Strukturoptimierung
Es wurde eine Strukturoptimierung für die zu synthetisierenden Komplexe durchgeführt und diese mit eventuell literaturbekannten Strukturen verglichen. Die optimierten Strukturen für die U(IV)-Komplexe mit iPr2Im- und Dipp2Im-Liganden (16 und 17) sind in Abbildung 12 dargestellt. Ein Vergleich dieser Strukturen mit den analogen bekannten Übergangsmetall- Komplexen mit Zr(IV) und Hf (IV)von NIEHUES et al. bzw. dem U(IV)-Komplex von GARDNER
aus dem Jahr 2009 zeigt dabei gute Übereinstimmung der Strukturen.[25,26]
Abbildung 12 Darstellung der optimierten Struktur des [U(iPr2Im)2Cl4]-Komplexes und des bekannten analogen Zr-Komplexes (oben links bzw. rechts).[25] Darstellung der optimierten Struktur des [U(Dipp2Im)2Cl4]-Komplexes und des von GARDNER publizierten Komplexes (unten links bzw. rechts).[26]
iPr2Im als Ligand
Dipp2Im als Ligand
16 12
17 13
11
3.2 Charakterisierung von Uran-Komplexen mit 1,3- Diisopropylimidazol-2-yliden
Einkristallröntgendiffraktometrie
[U
IV(
iPr
2Im)
2(N(SiMe
3)
2)Cl
3]-Komplex (18)
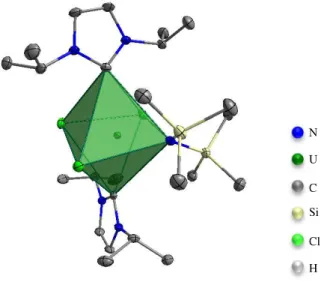
Bezüglich der Synthesen mit UCl4 konnten durch Diffusion von Benzol in eine THF- Mutterlösung grüne Kristalle gezüchtet werden. Dabei handelt es sich um einen [UIV(iPr2Im)2(N(SiMe3))2Cl3]-Komplex (18), dessen Molekülstruktur in Abbildung 13 dargestellt ist.
Abbildung 13 Molekülstruktur des synthetisierten [UIV(iPr2Im)2(N(SiMe3)2)Cl3]-Komplexes (18).
Hierbei wird das Metallzentrum von drei Chloroliganden, zwei iPr2Im- und einem Bis(trimethylsilyl)amido-Liganden umgeben. Durch die sechs Liganden ergibt sich eine oktaedrische Koordinationssphäre, die jedoch durch die sterischen und elektronischen Einflüsse des Amido-Liganden verzerrt ist (Abbildung 14). Des Weiteren ist in der Komplexstruktur ein Benzolmolekül (C25-C27) eingelagert, das jedoch nicht an das Metallzentrum koordiniert.
18
N U C Si Cl H
U1 Cl2
Cl1 Cl3
C1 N5
N1 N2
C3 C2 C4 C10
Si5 Si6
C26 C25 C27
12
Abbildung 14 Darstellung der oktaedrischen Koordinationssphäre des Uranzentrums. Wasserstoffatome sind zur Übersichtlichkeit weggelassen.
Die durchschnittliche U–Cl-Bindungslänge von 2,632 Å befindet sich in einem ähnlichen Bereich wie im es im [UCl6]2− Ion mit einem durchschnittlichen Abstand von 2,627 Å der Fall ist.[30] Allerdings ist die durchschnittliche U–Cl-Bindungslänge in diesem Fall nicht repräsentativ, da es sich bei dem Koordinationspolyeder um ein verzerrtes Oktaeder handelt und sich damit der längste und der kürzeste U–Cl Abstand um 0,33 Å unterscheiden. Betrachtet man den kürzesten Abstand, so ist auffällig, dass es sich dabei um das Chloratom handelt, welches trans zu dem Amido-Liganden steht. In diesem Zusammenhang könnte eine Veränderung der elektronischen Umgebung des Chloroliganden stehen. Das Stickstoffatom des Amido-Liganden kann aufgrund des freien Elektronenpaares Elektronendichte in Richtung des Metallzentrums verlagern. Die zusätzliche Elektronendichte könnte damit zu einer Verkürzung der Bindung zum Cl2 führen. Dieses Phänomen wird als inverser trans-Einfluss bezeichnet.
Dabei stehen stark donierende Liganden trans zueinander und beeinflussen sich gegenseitig, was zu einer Verkürzung der Bindungen führen kann.[31] Wird der U–N-Abstand beleuchtet, so ist dieser mit 2,242 Å sogar noch kürzer als im [U(N(SiMe3)2)4]-Komplex, in dem er bei 2,297 Å liegt.[32] Damit muss mit dem Chloroliganden ebenfalls eine Wechselwirkung stattfinden, die den Bindungsabstand beeinflusst. Wird der U–N-Abstand mit jenen in {U-μ- Cl[N(SiMe3)2]2[=N(SiMe3)]}2verglichen,so zeigt sich Übereinstimmung der experimentell bestimmten 2,242 Å zu den dort bestimmten 2,243 Å der N(SiMe3)2-Liganden.[33] Damit kann postuliert werden, dass der Amido-Ligand nicht nur die Cl-Bindungsabstände beeinflusst, sondern, dass der U–N-Abstand auch durch das Vorhandensein von Chloriden beeinflusst wird.
Ein Vergleich mit der theoretisch berechneten Struktur zeigt, dass die experimentell bestimmten Cl-Bindungslängen größer sind, als die berechneten 2,604 Å. Übereinstimmend damit sind die U–C-Bindungsabstände im berechneten Komplex mit 2,562 Å um ca. 0,1 Å kürzer als in der experimentell bestimmten Molekülstruktur. Denkbar ist hierbei, dass die Carben-Liganden durch die Trimethylsilyl-Gruppen weiter vom Uran weggedrückt werden, um repulsive
N U C Si Cl H
13 Wechselwirkungen des Amido-Liganden mit den Isopropylgruppen zu vermeiden. Zumeist können die Unterschiede zwischen den theoretischen und experimentellen Daten auch in intermolekularen Wechselwirkungen, die die Theorie nicht berücksichtigen kann, begründet sein.
Tabelle 2 Bindungslängen zwischen U1 und allen unmittelbaren Nachbaratomen.
Atom 1 Atom 2 Abstand d1,2 [Å]
U1 Cl1 2,630
Cl2 2,617
Cl3 2,650
N5 2,242
C1 2,658
C10 2,667
Im berechneten Komplex 16 bilden die beiden Carben-Kohlenstoffe und das Uran einen Winkel von 180°, während man in der Molekülstruktur 18 einen C–U–C Winkel von 165° findet. Diese signifikante Winkelverkleinerung kann mit der Koordination des Amido-Liganden erklärt werden. Durch die sterisch anspruchsvollen Trimethylsilyl-Gruppen werden die Carbene aus ihrer Ebene hinausgedrückt, um Wechselwirkungen zwischen den Methyl- und Isopropylgruppen zu minimieren. Das äußert sich auch darin, dass die Carben-Liganden nicht wie im berechneten Komplex in einer Ebene liegen, sondern um 88,8° zueinander verdreht sind.
Der N(SiMe3)2-Ligand ist deshalb auch um 56,8° gegenüber der C–U–C-Ebene geneigt, um die Trimethylsilyl-Gruppen zwischen den Chloroliganden und Isopropylgruppen zu arrangieren.
Der Amido-Ligand drückt ebenfalls die Chloroliganden aus ihrer Ebene, sodass nicht mehr der theoretische Winkel von 180°, sondern einen Winkel Cl1–U1– Cl3 von 170,7° erreicht wird.
Ein Vergleich der Cl2–U1–Cl3- bzw. Cl2–U1–C1-Winkel zeigt, dass diese zum einen nahezu identisch sind und zum anderen, dass sich die Winkel im Komplex mit 86° bzw. 87° denen im berechneten Komplex (87°) gleichen. Allerdings finden sich im berechneten Komplex zwei unterschiedliche Winkel zwischen den vier Chloroliganden. Die zwei auf einer Seite der Ligandebene schießen den kleineren Winkel von 87° ein, während die zwei, die sich jeweils auf unterschiedlichen Seiten der Ligandebene befinden, einen größeren Winkel von ca. 92°
einschließen. Erklärt werden könnte dies mit dem Bestreben der Vermeidung von repulsiven Interaktionen mit den Isopropylsubstituenten.
Tabelle 3 Ausgewählte Bindungswinkel im [UIV(iPr2Im)2(N(SiMe3)2)Cl3]-Komplex.
Atom 1 Atom 2 Atom 3 ∠1,2,3 [°]
C1 U1 C10 165,7
Cl1 U1 Cl3 170,7
Cl2 U1 N5 174,1
Si6 N5 Si5 116,1
C1 U1 Cl3 82,0
C1 U1 Cl1 91,7
C1 U1 N5 98,5
14
Abbildung 15 Visualisierung der unterschiedlichen Cl–U–Cl-Winkel im berechneten [U(iPr2Im)2Cl4]-Komplex 16 (links) und in 18 (rechts).
Im bereits bekannten [U(Dipp2Im)2Cl4]-Komplex 17 zeigt sich ebenfalls, dass die Chloro- Liganden nicht in einer Ebene liegen, sondern einen Winkel Cl1–U1–Cl3 von 165,8°
aufspannen. Diese starke Abweichung von den erwarteten 180° wird in diesem Fall jedoch mit den Cl-π-Wechselwirkungen zwischen den Liganden und den Phenylringen begründet, die im Fall von 18 nicht existieren.[26]
Abbildung 16 Darstellung der kurzreichweitige Wechselwirkung (rot) zwischen benachbarten Molekülen des [UIV(iPr2Im)2(N(SiMe3)2)Cl3]-Komplexes.
Werden nun benachbarte Moleküle betrachtet (Abbildung 16), so existieren CHX- Wechselwirkungen C3–H3∙∙∙Cl2 zwischen zwei benachbarten Molekülen, mit einer Länge von 2,85 Å und einem Winkel von 118°. Laut einer Einteilung von JEFFREY fällt diese Interaktion damit in den Bereich von 2,5-3,2 Å und kann damit als moderat und hauptsächlich elektrostatischer Natur eingestuft werden.[34]
Wird das Augenmerk auf die Packung der Moleküle gelegt, so wird aus Abbildung 17 deutlich, dass die Trimethylsilyl-Gruppen von vier Molekülen zueinander gerichtet sind, wobei sich in deren Mitte ein Benzolmolekül (orange) befindet. Damit zeigen die Carben-Liganden zueinander und sind somit in der Lage die beschriebenen Wechselwirkungen auszubilden. Die Benzolmoleküle bilden wider Erwarten keinerlei Wechselwirkungen zu den Komplex-
N U C Si Cl H 92° 87°
Carben- Ligand- Ebene
85,9°
86,9°
N U C Si Cl H H3
Cl2 C3
15 Molekülen oder untereinander aus und füllen damit nur die Hohlräume in der Struktur.
Aufeinanderfolgende Benzolmoleküle sind jeweils um 90° verkippt, sodass jede zweite Reihe dieselbe Ausrichtung besitzt. Die Zentroide der übereinanderliegenden Benzolmoleküle in Abbildung 17 besitzen einen Abstand von 11,777 Å.
Abbildung 17 Packung der [UIV(iPr2Im)2(N(SiMe3)2)Cl3]-Komplex-Moleküle im Kristall. Zur besseren Übersichtlichkeit sind die Benzolmoleküle orange dargestellt und Wasserstoffatome weggelassen.
Ob es sich bei dieser Struktur auch um die in Lösung stabile Spezies handelt, ist zweifelhaft.
Indizien hierfür sind zum einen die Farbänderung, welche beim Lösen der Substanz in Acetonitril auftritt, zum anderen entsprechen die bei der NMR-Untersuchung gefundenen Signale nicht denen, die für eine solche Struktur erwartet werden würden (Kapitel 3.2.3).
[U(
iPr
2ImH)
2(=N(SiMe
3))Cl
5]-Komplex (19)
Neben der genannten Struktur konnten ebenfalls aus einer braunen Komplexlösung, die
iPr2ImHCl, LiHMDS und UCl4 enthielt, hellgrüne plättchenförmige Kristalle gewonnen werden. Eine röntgenographische Untersuchung derer lieferte folgende Molekülstruktur.
a c b
N U C Si Cl H
16
Abbildung 18 Molekülstruktur des synthetisierten[U(iPr2ImH)2(=N(SiMe3))Cl5]-Komplexes (19).
Aus Abbildung 18 wird ersichtlich, dass das U-Zentrum wie in 18 oktaedrisch umgeben ist und dass neben fünf Chloroliganden zusätzlich ein Trimethylsilylimidoligand an das Metall koordiniert. Dessen Ursprung liegt in der eingesetzten Base, wobei sich diese während der Reaktion zersetzt haben muss. Wie genau das geschehen ist, ist zum jetzigen Zeitpunkt noch nicht geklärt. Das Uran ist das Zentrum des Komplexanions, dessen negative Ladung durch zwei protonierte und damit positiv geladene Imidazoliumionen ausgeglichen wird.
Wird das Augenmerk zunächst auf die U–N-Bindungslänge gelegt, so wird deutlich, dass diese mit 2,089 Å deutlich kürzer ist, als im [U(N(SiMe3)2)4] Komplex mit 2,297 Å.[32] Verglichen mit dem {U-μ-Cl[N(SiMe3)2]2[=N(SiMe3)]}2-Komplex, in dem eine U–N-Bindungslänge von 2,081 Å zu finden ist, lässt sich eine gute Übereinstimmung der Längen feststellen.[33] Dabei bezieht sich der genannte Wert auf die U–N-Doppelbindung, die in der literaturbekannten Verbindung zu finden ist. Der verhältnismäßig kurze U–N-Abstand deutet auf eine U–N- Doppelbindung hin. Diesbezüglich haben theoretische Berechnungen eine U–N Bindungsordnung von 1,8 ergeben, was insgesamt zu dem Schluss der Existenz einer U–N Doppelbindung führt. Mit dieser Erkenntnis und einer Betrachtung der groben Formalladungen gelangt man zu dem Schluss, dass das Uran in diesem Komplex die Oxidationsstufe +5 besitzen muss. Werden die U–Cl Bindungsabstände betrachtet (Tabelle 4), so sind die zu Cl3, Cl4 und Cl5 in einem sehr ähnlichen Bereich. Die Bindungen zu Cl1 und Cl2 hingegen sind deutlich kürzer, weisen aber zum Imidoliganden keine besondere Stellung auf. Verglichen mit dem U(IV) Komplex 18 sind die U–Cl Abstände in diesem Komplex etwas länger, was mit der veränderten Ligandumgebung erklärt werden kann. Auch die N–Si-Bindung ist mit 1,61 Å deutlich kürzer als im [U(N(SiMe3)2)4] mit ca. 1,75 Å.[32]
N U C Si Cl H Cl1
Cl3 Cl2 Cl4
Cl5 N1 Si1
C1 N2
N3 C3
C2 C5
C4
U1
19
17
Tabelle 4 Ausgewählte Bindungslängen im [U(iPr2ImH)2(=N(SiMe3))Cl5]-Komplex.
Atom 1 Atom 2 Abstand d1,2 [Å]
U1 Cl1 2,656
Cl2 2,663
Cl3 2,689
Cl4 2,685
Cl5 2,680
N1 2,089
N1 Si1 1,610
Die Chloroliganden Cl2 und Cl4 sowie Cl5 und der Imidoligand stehen sich nahezu direkt gegenüber, was durch Bindungswinkel ca. 180° ausgedrückt wird. Mit 173° lässt sich eine nahezu lineare U–N–Si Bindung verzeichnen.
Tabelle 5 Ausgewählte Bindungswinkel in 19.
Atom 1 Atom 2 Atom 3 ∠1,2,3 [°]
Si1 N1 U1 173,1
N1 U1 Cl5 178,8
Cl4 U1 Cl2 172,6
Ein Blick auf die Packung der Moleküle (Abbildung 17) zeigt, dass sich eine schichtartige Zickzack-Struktur ausbildet. Der Trimethylsiliylimidoligand weist dabei in Richtung der a- Achse.
Abbildung 19 Packung von [U(iPr2ImH)2(=N(SiMe3))Cl5] (19).
Alle durch rote Linien symbolisierten intermolekularen Cl∙∙∙H–C Wechselwirkungen in Abbildung 20 liegen in einem Bereich von 2,6-2,9 Å. Die Winkel unterscheiden sich jedoch deutlich. Die Wechselwirkungen, die vom Cl2 ausgehen weisen Winkel von 160-175° auf, sind aufgrund ihrer Länge jedoch eher als moderat einzustufen. In einem ähnlichen Längenbereich liegen auch die Interaktionen, in die Cl3 involviert ist. Allerdings lassen sich hier Winkel um 130-140° finden. Bei der Interaktion Cl4∙∙∙H3–C3 ist ein Winkel von ca. 157° und eine Länge von 2,67 Å zu finden. Damit sind auch diese Wechselwirkungen, genau wie die Cl4∙∙∙H1–C1 Interaktion mit 135° und 2,69 Å als moderat und größtenteils elektrostatischer Natur einzuschätzen.[34]
N U C Si Cl H c
a
18
Abbildung 20 Intermolekulare Wechselwirkungen in der Struktur des [U(iPr2ImH)2(=N(SiMe3))Cl5]-Komplexes.
Infrarot-Spektroskopie
Das pulverförmige Produkt aus der Umsetzung von 9a mit UCl4 wurde ebenso wie das reine Ligandsalz mittels ATR-FT-IR-Spektroskopie untersucht. Das isolierte freie Carben wurde in Form eines braunen Öls gemessen. Mit Hilfe der theoretisch berechneten IR-Spektren des freien Carbens und des erwarteten U(IV)-Komplexes 16 soll durch einen Vergleich die Bandenzuordnung und die Interpretation bzw. der Nachweis des Komplexes via IR- Spektroskopie gelingen. In Abbildung 21 sind genannte Spektren dargestellt. Die im Zuge der Spektrenauswertung beschriebenen Molekülschwingungen sind im Anhang visualisiert.
N U C Si Cl H Cl1
Cl2 Cl3
Cl4 H1
H1 H3
H2 C3 C2 C9
H9 H7
C7 H3 C3
19
Abbildung 21 IR-Spektrum des synthetisierten bulk-Materials [U(iPr2Im)2Cl4] (dunkelgrün), des freien Carbens (hellgrün) und des Imidazoliumsalzes (rot) im Vergleich zu den theoretisch berechneten (bezeichnet mit ber.) Spektren des freien Carbens (orange) und des U(IV)-Komplexes (blau).
In beiden berechneten Spektren lassen sich Valenzschwingungen der CH3- und CH-Gruppen der Isopropylreste im Wellenzahlbereich um 3000 cm-1 wiederfinden. In jenem Bereich treten ebenfalls Banden in den gemessenen Spektren auf. Problematisch ist jedoch, dass, falls in den synthetisierten Produkten protonierter Ligand sein sollte, die entscheidende CH-Schwingung am C1 nicht identifizierbar ist, da diese auch um 3000 cm-1 auftritt, was durch die breite Bande im Spektrum des reinen Ligandsalzes deutlich wird. Der berechnete [U(iPr2Im)2Cl4]-Komplex zeigt bei ca. 1360 cm-1 eine breite und intensive Bande, welche durch die asymmetrische Valenzschwingung N–C1–N und der scissoring Deformationsschwingung der CH3-Gruppen (Schulter bei ca. 1410 cm-1) hervorgerufen wird. Weil bei dieser Schwingung das Carben-C involviert ist, kann diese Bande als Markerbande bezeichnet werden, da sie für die Verbindung charakteristisch ist und von der Bindung zu einem Metallzentrum in ihrer Lage, verglichen zum freien Carben, beeinflusst werden sollte. Die entsprechende Bande liegt im Spektrum des freien Carbens bei ca. 1100 cm-1. Durch die Bindung zum Metall ist demnach mehr Energie nötig, um die Schwingung anzuregen, weshalb sie bei höheren Wellenzahlen auftritt. Allerdings liegt im Wellenzahlbereich der Markerbande um 1360 cm-1 im Spektrum des freien Carbens die Bande der asymmetrischen Valenzsschwingung C(iPr)–N–C(Methylen). Dadurch wird die Bandenzuordnung erschwert, da eine eindeutige Unterscheidung nicht mehr möglich ist. Der berechnete Komplex zeigt bei 1160 cm-1 eine sehr intensive Bande, welche jedoch die rocking- Deformationsschwingung der Methylenkohlenstoffe darstellt, die durch die Koordination zum Metall nicht beeinflusst werden sollte. Im Wellenzahlbereich um und unter 1000 cm-1 finden sich strukturanalytisch weniger interessante Deformationsschwingungen der CH-Guppen der
1000 1500
2000 2500
3000 3500
Transmission [%]
ν [cm-1]
iPr2Im ber.
iPr2Im
iPr2ImHCl
[U(iPr2Im)2Cl4] ber.
[U(iPr2Im)2Cl4]
20 übereinstimmen. Teilweise Übereinstimmungen sind auch zu den Spektren des Ligandsalzes und des isolierten Carbens sichtbar. Betrachtet man das berechnete und gemessene Spektrum des freien Carbens, so wird auch hier gute Übereinstimmung ersichtlich. Das berechnete Spektrum kann damit als Hilfe für die Bandenzuordnung verstanden werden. Diesbezüglich kann gesagt werden, dass in den Spektren des Produktes und des freien Liganden die um 3000 cm-1 und 600 cm-1 erwarteten Valenz- und Deformationsschwingungen der Isopropylreste verzeichnet werden können. Dies ist auch der Fall im Spektrum des Ligandsalzes, wobei zusätzlich Schwingungen der CH-Gruppe am C1 des Imidazoliumrings beteiligt sind. Die sehr intensive Bande um 1200 cm-1 kann der Deformationsschwingung der Methylenkohlenstoffe zugeordnet werden, wobei sie im Vergleich zum Spektrum des bulk-Materials minimal zu höheren Wellenzahlen verschoben ist. Besonders auffallend ist, dass das Dublett um 1360 cm-1 im Spektrum des freien Carbens im Vergleich zum Spektrum des Ligandsalzes um fast 100 cm-1 auf ca. 1270 cm-1 verschoben ist. Neben der Zuordnung dieser Banden zur Valenzsschwingung C(iPr)–N–C(Methylen) des freien Carbens oder der asymmetrischen Valenzschwingung N–C1–N des Uran-Carben-Komplexes, besteht zudem die Möglichkeit, dass das Dublett durch eine Deformationsschwingung der CH3-Gruppen erzeugt worden ist.
Bei einer Kettenverzweigung aliphatischer Strukturen, wie es bei Isopropylgruppen der Fall ist, kann es zu einer solchen charakteristischen Aufspaltung mit gleichen Intensitäten kommen.[35]
Dieses Dublett lässt sich im Spektrum des synthetisierten Produktes aus der Umsetzung mit UCl4 ebenfalls bei ca. 1360 cm-1 finden. Dessen Ursprung lässt sich aufgrund der beschriebenen vielfältigen Möglichkeiten zwar nicht eindeutig festlegen, es ist jedoch möglich, die Wahrscheinlichkeiten der genannten Optionen abzuwägen. Begründet mit der ausgeprägten Dublettstruktur und der gleichen Intensitäten der Bandeteile, ist es am wahrscheinlichsten, dass es sich um die Deformationsschwingung der Methylgruppen handelt.
Des Weiteren konnte ein IR-Spektrum der aus einer Synthese bei Raumtemperatur erhaltenen grünen Kristalle des [UIV(iPr2Im)2(N(SiMe3)2)Cl3]-Komplexes 18 aufgenommen werden. In Abbildung 22 sind die entsprechenden Spektren dargestellt.
21
Abbildung 22 IR-Spektren des pulverförmigen bulk-Materials (grün) und des kristallinen [UIV(iPr2Im)2(N(SiMe3)2)Cl3]-Komplexes 18 (violett) im Vergleich zum Spektrum des freien Carbens (hellgrün) und theoretisch berechneten (bezeichnet mit ber.) Spektrum des U(IV)-Komplexes 16 (blau).
Zunächst wird deutlich, dass sich die Spektren der synthetisierten Produkte unterscheiden. Das könnte bspw. daran liegen, dass in der Molekülstruktur des [UIV(iPr2Im)2(N(SiMe3)2)Cl3]- Komplexes nur drei Chloroliganden, aber zusätzlich noch ein Trimethylsilylamido-Ligand an das Metall koordinieren. Die damit erheblich abweichende Struktur gegenüber der für die Rechnung angenommene, könnte so die Unterschiede erklären, da im synthetisierten Komplex andere Schwingungen möglich sind. Zudem ist eine teilweise Übereinstimmung zwischen den Spektren des freien Carbens 9a und des kristallinen Komplexes zu verzeichnen. Die Bandenzuordnung, die bzgl. des freien Carbens (grünes Spektrum in Abbildung21) getroffen wurde, ist aufgrund der großen Ähnlichkeit zum Spektrum des [UIV(iPr2Im)2(N(SiMe3)2)Cl3]- Komplexes (violettes Spektrum in Abbildung 22) in diesem ebenfalls gültig. Die erhebliche Intensitätssteigerung der Banden um 3000 cm−1 im violetten Spektrum kann durch das Vorhandensein von sechs weiteren Methylgruppen erklärt werden, deren Valenzschwingungen ebenfalls zur Intensität der um 3000 cm-1 gelegenen Bande beitragen. Unerwartet ist die Verschiebung der um 1660 cm-1 befindliche Bande im hellgrünen Spektrum um fast 100 cm-1 zu 1550 cm-1 in den anderen Spektren (schwarze Pfeile Abbildung 22). Diese sehr schwache Bande wird der Valenzschwingung der Methylengruppen im Imidazoliumring zugeordnet und sollte durch die Koordination zu einem Metall nicht stark beeinflusst werden, da keine direkte Bindung besteht. Außerdem kann nicht gesagt werden, durch welche Schwingungen die Bande bei ca. 850 cm-1 (bulk Material, dunkelgrünes Spektrum) hervorgerufen wird, da weder im Spektrum des gemessenen Kristalls, noch im berechneten Spektrum des Komplexes im besagten Bereich eine Bande zu sehen ist.
1000 1500
2000 2500
3000 3500
Transmission [%]
ν [cm-1]
iPr2Im
[U(iPr2Im)2Cl4] ber.
[U(iPr2Im)2(N(SiMe3)2)Cl3] Kristall
[U(iPr2Im)2Cl4]
22 ergänzende Methode, um die Ergebnisse aus anderen strukturaufklärenden Methoden, wie NMR oder SC-XRD zu untermauern.
NMR-Spektroskopie
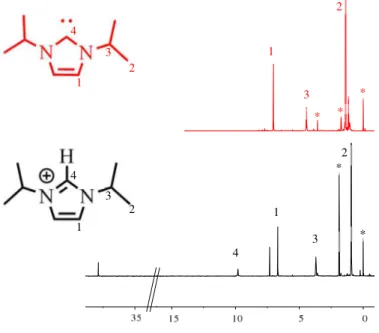
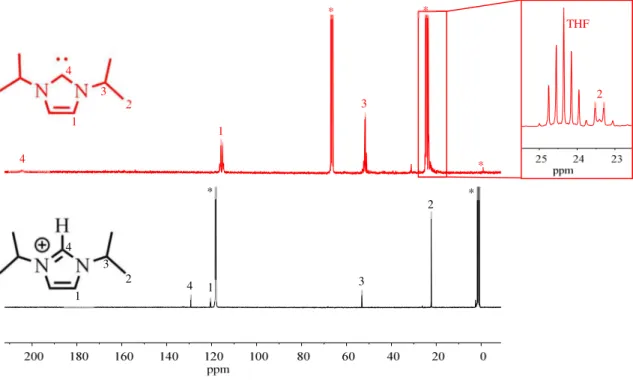
Die bei der Umsetzung von UCl4 mit iPr2ImHCl und LiHMDS in MeCN erhaltene braune Komplexlösung, aus der auch die grünen Kristalle von 18 gewonnen wurden, wurde nach dem Eintrocknen in deuteriertem MeCN aufgenommen und via NMR untersucht. Dabei konnten im
13C-Spektrum alle Signale und im 1H-Spektrum (Abbildung 23 und Abbildung 24) fast alle Signale, außer jene bei 38 ppm und ca. 7 ppm, zugeordnet werden. Des Weiteren sind kaum Verunreinigungen zu erkennen, was für eine gute Qualität der Probe spricht.
Abbildung 23 1H-NMR Spektren des aus der Umsetzung von UCl4 mit iPr2ImHCl und LiHMDS erhaltenen Produktes (schwarz, MeCN-d3) und des freien Carbens (rot, THF-d8). Mit * sind TMS- und Lösungsmittelpeaks markiert.
Die Signale 2 und 3 gehören zu den Isopropylsubstituenten und liegen, im Vergleich zum freien Carben, im erwarteten Bereich. Dies bedeutet jedoch, dass diese Gruppen keinen paramagnetischen Einfluss erfahren. Dies ist auch für 1 der Fall, das aufgrund der 2D Spektren den Brückenatomen zugeordnet werden konnte und auch in einem ähnlichen Bereich wie bei dem freien Carben zu finden ist. Das Signal 4 kann dem Proton am Carben-Kohlenstoff zugeordnet werden und beweist damit, dass in der untersuchten Probe der Ligand in seiner protonierten Form vorliegt. Signal 4 ist im Vergleich zum roten Spektrum hochfeldverschoben, was durch das zusätzliche Proton am Carben-Kohlenstoff erklärt werden kann, da es die
4
1
2 3
3 2
1
1
2 3 4
4 1
2
3
*
*
*
*
*
23 elektronische Umgebung an jenem verändert. Interessant ist das Signal bei ca. 38 ppm im 1H- Spektrum, da es für ein Protonenspektrum sehr stark verschoben ist und gesagt werden kann, dass es einen paramagnetischen Einfluss erfahren hat. In den aufgenommenen 2D-Spektren ist zwar ein Kreuzpeak passend zu dem Signal bei ca. 38 ppm zu finden, allerdings konnte im 13C- NMR Spektrum an entsprechender Stelle (ca. 80 ppm) kein Signal gefunden werden (Abbildung 24). Möglicherweise gibt es ein Signal, das jedoch durch ungünstige Relaxation so stark verbreitert ist, dass es im 1D 13C-Spektrum nicht zu sehen ist. Ein paramagnetischer Einfluss, der zu einer Verbreiterung beitragen kann, ist ebenso möglich. Da im 2D-Spektrum die heteronukleare Korrelation zwischen den empfindlichen 1H-Kernen und den 13C-Kernen gemessen wird, ist dort ein Kreuzpeak sichtbar, obwohl im 13C-Spektrum kein Signal gefunden werden konnte. Denkbar ist, dass das Signal bei 38 ppm aus der Koordination von Lösungsmittelmolekülen oder eines (Me3Si)2N-Restes zum Uran herrührt. Wäre dies der Fall, so würde eine hohe chemische Verschiebung, hervorgerufen durch den paramagnetischen Einfluss des Urans, für das Lösungsmittel- und (Me3Si)2N-Signal auftauchen. Insgesamt deuten die Ergebnisse der NMR-Spektroskopie auf eine Uran-Spezies hin, bei der es sich jedoch wahrscheinlich nicht um den [UIV(iPr2Im)2(N(SiMe3)2)Cl3]-Komplex 18 handelt, da für diese Struktur andere Signale zu erwarten wären. Dadurch, dass die Signale des Liganden eine Protonierung desselben bestätigen und nicht verschoben sind, kann angenommen, dass das Gleichgewicht der Komplexbildungsreaktion im zeitlichen Mittel auf der Seite der Edukte liegt.
Es könnte eventuell ein schneller Austausch zwischen den Komplexmolekülen und einer anderen U-Spezies stattfinden, sodass der Komplex nicht detektiert werden kann.
Abbildung 24 13C-NMR Spektren des aus der Umsetzung von UCl4 mit iPr2ImHCl und LiHMDS erhaltenen Produktes (schwarz, MeCN-d3) und des freien Carbens (rot, THF-d8). Mit * sind TMS- und Lösungsmittelpeaks markiert.
1 3 2 4
4
3 1
2
3
2
4 1 4
1
2 3
*
* *
*
THF
*
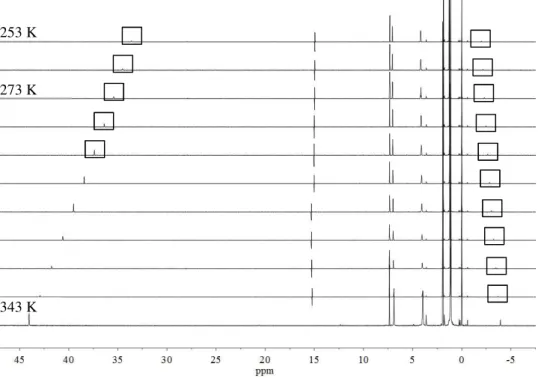
24 Tieffeldverschiebung mit steigender Temperatur erfährt. Dies könnte beispielsweise mit einem paramagnetischen Einfluss erklärt werden. Bei einem weiteren Signal bei ca. −2 ppm (253 K) ist ein ebenfalls ein Einfluss der Temperatur sichtbar. Mit steigender Temperatur tritt hier eine Hochfeldverschiebung auf. Was die Ursache hierfür ist und ob es sich dabei eventuell um eine weitere Spezies handeln könnte, ist noch nicht geklärt.
Abbildung 25 Temperaturreihe des aus der Umsetzung von UCl4 mit iPr2ImHCl und LiHMDS erhaltenen Produktes von 253-343 K.
3.3 Charakterisierung von Uran-Komplexen mit 1,3- Bis(2,6-diisopropylphenyl)imidazol-2-yliden
Einkristallröntgendiffraktometrie
1,3-Bis(2,6-diisopropylphenyl)imidazol-2-yliden (9b)
Durch Verdunstung einer Lösung von 9b in THF konnten farblose, stäbchenförmige Kristalle gewonnen werden, bei denen es sich um das freie Dipp2Im-Carben 9b handelt (Abbildung 26).
253 K
343 K 273 K
25
Abbildung 26 Molekülstruktur des freien Carbens 1,3-Bis(2,6-diisopropylphenyl)imidazol-2-yliden (9b).
Die asymmetrische Einheit enthält nur das elektrisch neutrale Carbenmolekül.
Werden die experimentell bestimmten Bindungslängen, mit denen der Literatur verglichen, so zeigt sich, dass die C–N-Bindungslängen mit durchschnittlich 1,374 Å gut zum bekannten Wert von 1,367 Å passen. Ähnlich gute Übereinstimmung zeigt der Abstand zwischen N1–C2 mit 1,384 Å bzw. N2–C3 von 1,392 Å zu durchschnittlich 1,392 Å. Der C2–C3-Abstand ist mit 1,350 Å hingegen etwas länger als die von NIEHUES et al. bestimmten 1,335 Å.[36] Als nahezu identisch zu den Literaturwerten können die Bindungswinkel im Molekül vermerkt werden.
Der Winkel N1–C1–N2 liegt mit 101,0° nur unwesentlich unter dem Literaturwert von 101,4°.
Die experimentell bestimmten Winkel C1–N1–C2 (113,7°) und N1–C2–C3 (105,9°) passen ebenso jeweils zu den literaturbekannten werden von 113,0° und 106,3°.[36]
In Abbildung 27 ist die Packung der Moleküle im Kristall dargestellt. Sichtbar ist, dass die Phenylringe nahezu in eine Richtung zeigen. Dadurch weisen die Isopropyl-Gruppen ebenfalls in eine Richtung, sodass repulsive Wechselwirkungen vermieden werden. Durch die aufeinander gerichteten Phenylringe sind Wechselwirkungen zwischen diesen möglich. Die in der Packung präsenten intermolekularen Wechselwirkungen sind in Abbildung 28 als rote Linien eingezeichnet.
Abbildung 27 Packung der 1,3-Bis(2,6-diisopropylphenyl)imidazol-2-yliden-Moleküle im Kristall.
N H C C1
C2 C3 N2 N1
C4 C16
9b
a c
N C H
26 2,682 Å und es wird ein C1–H2–C2 Winkel von 150,4° ausgebildet. Aufgrund dessen kann diese Wechselwirkung nach JEFFREY als moderat und hauptsächlich elektrostatischer Natur eingestuft werden.[34] Die Zentroide von übereinander liegenden Phenylringen sind 5,774 Å voneinander entfernt, wodurch keine π-π-Wechselwirkungen ausgebildet werden können (hellblaue, gepunktete Linie in Abbildung 28). Als Vergleichswert wird dafür der Abstand der π-π-Wechselwirkung in Porphyrinen herangezogen, der 3,4-3,6 Å beträgt.[37]
Abbildung 28Intermolekulare Wasserstoffbrückenbindungen zwischen 1,3-Bis(2,6-diisopropylphenyl)imidazol- 2-yliden-Molekülen (rote Strichlinie).
[HDipp
2Im]
2[UCl
6]∙MeCN (20)
Aus einer Synthese von UCl4 und Dipp2ImHCl in MeCN konnten mit Hilfe des Diffusionsansatzes mit Et2O hellgrüne, quaderförmige Kristalle gezüchtet werden. Durch eine röntgenographische Untersuchung konnte die Struktur des entstandenen Salzes [HDipp2Im]2[UCl6]∙MeCN (20) aufgeklärt werden (Abbildung 29).
C1 C3
H3
N C H
27
Abbildung 29 Molekülstruktur des synthetisierten [HDipp2Im]2[UCl6]∙MeCN-Komplex.
Es handelt es sich dabei um [UCl6]2−-Oktaeder, deren negative Ladungen von zwei positiv geladenen, protonierten Ligandmolekülen ausgeglichen werden. Des Weiteren befindet sich ein MeCN-Molekül in der Molekülstruktur. Die U–Cl-Bindungslängen sind in Tabelle 6 dargestellt. Die U1–Cl1- und U1–Cl2-Abstände sind nahezu identisch, was durch die äquatoriale Lage dieser Atome zu erwarten ist. Ein Vergleich zur Literatur zeigt zunächst gute Übereinstimmung zu den angegebenen 2,621 Å bzw. 2,623 Å für Chloroliganden in äquatorialer Ebene und 2,627 Å für die horizontalen Chloroliganden.[38] Übereinstimmend findet man auch hier zwei leicht unterschiedliche U–Cl-Abstände in der äquatorialen Ebene, die um 0,003 Å voneinander abweichen.
Tabelle 6 U–Cl-Abstände in der Kristallstruktur des [HDipp2Im]2[UCl6]∙MeCN-Komplexes.
Atom 1 Atom 2 Abstand d1,2 [Å]
U1 Cl1 2,626
Cl2 2,633
Cl3 2,629
C1 N1
N2
1,312 1,312
N1 C2 1,490
N1 C4 1,442
N2 C3 1,497
N2 C16 1,448
C2 C3 1,555
Im Vergleich zu 13, publiziert von GARDNER, MCMASTER und LIDDLE, sind alle U–Cl- Abstände allerdings länger als die dortigen durchschnittlichen 2,571 Å.[26] Eine mögliche Begründung ist die fehlende Bindung zum Carben. Allerdings sind die U–Cl-Abstände kürzer als in 18, wobei in diesem Fall die Koordination des Amido-Liganden mitberücksichtigt werden muss. Im Vergleich zum freien Carben kommt es zur Aufweitung des N1–C1–N2-Winkels auf
U1 Cl1
Cl3 Cl2
N2 C1 C3 C16
N1 C4
C2
20
N U C Cl H N1
28 hinweist. Demnach muss es während der Reaktion zu einer Reduktion des Imidazoliumrings gekommen sein. Die C–N-Abstände sind mit 1,312 Å zudem deutlich kürzer als die 1,374 Å im freien Carben. Ein signifikanter Unterschied ist ebenfalls bei der Bindung der Stickstoffatome zu den Kohlenstoffatomen der Imidazoliumbrücke zu verzeichnen. In 20 liegt dieser Abstand durchschnittlich bei 1,494 Å, wohingegen in 9b ein Wert von 1,388 Å zu finden ist. Insgesamt wird durch die Reduktion der Doppelbindung nicht nur der C2–C3 Abstand beeinflusst, sondern auch weitere Strukturparameter des Imidazoliumrings.
Die Phenylringe zweier direkt benachbarter Imidazoliumionen können zwar überlappen, jedoch ist der Zentroid-Abstand (hellblaue, gestrichelte Linie, Abbildung 30 rechts) mit 5,53 Å zu lang, als dass eine π-π-Wechselwirkung stattfinden könnte. Als Vergleichswert wird dafür der Abstand der π-π-Wechselwirkung in Porphyrinen herangezogen, der 3,4-3,6 Å beträgt.[37]
Tabelle 7 Ausgewählte Bindungswinkel im [HDipp2Im]2[UCl6]-Komplex.
Atom 1 Atom 2 Atom 3 ∠1,2,3 [°]
Cl1 U1 Cl2 90,9
Cl1 U1 Cl3 89,4
N1 C1 N2 114,1
N1 C2 C3 102,9
C1 N1 C2 110,2
C1 N1 C4 124,2
Abbildung 30 Packung der Moleküle im Kristall der Verbindung 20.
N U C Cl H b c c
a
![Tabelle 3 Ausgewählte Bindungswinkel im [U IV ( i Pr 2 Im) 2 (N(SiMe 3 ) 2 )Cl 3 ]-Komplex](https://thumb-eu.123doks.com/thumbv2/1library_info/4565386.1599813/17.892.107.795.984.1155/tabelle-ausgewählte-bindungswinkel-iv-pr-im-sime-komplex.webp)
![Abbildung 15 Visualisierung der unterschiedlichen Cl–U–Cl-Winkel im berechneten [U( i Pr 2 Im) 2 Cl 4 ]-Komplex 16 (links) und in 18 (rechts)](https://thumb-eu.123doks.com/thumbv2/1library_info/4565386.1599813/18.892.110.618.103.364/abbildung-visualisierung-unterschiedlichen-cl-cl-winkel-berechneten-komplex.webp)
![Abbildung 17 Packung der [U IV ( i Pr 2 Im) 2 (N(SiMe 3 ) 2 )Cl 3 ]-Komplex-Moleküle im Kristall](https://thumb-eu.123doks.com/thumbv2/1library_info/4565386.1599813/19.892.110.520.226.597/abbildung-packung-der-iv-sime-komplex-moleküle-kristall.webp)
![Abbildung 21 IR-Spektrum des synthetisierten bulk-Materials [U( i Pr 2 Im) 2 Cl 4 ] (dunkelgrün), des freien Carbens (hellgrün) und des Imidazoliumsalzes (rot) im Vergleich zu den theoretisch berechneten (bezeichnet mit ber.) Spektren des fr](https://thumb-eu.123doks.com/thumbv2/1library_info/4565386.1599813/23.892.123.674.121.480/abbildung-synthetisierten-materials-dunkelgrün-imidazoliumsalzes-vergleich-theoretisch-berechneten.webp)
![Abbildung 22 IR-Spektren des pulverförmigen bulk-Materials (grün) und des kristallinen [U IV ( i Pr 2 Im) 2 (N(SiMe 3 ) 2 )Cl 3 ]-Komplexes 18 (violett) im Vergleich zum Spektrum des freien Carbens (hellgrün) und theoretisch berechneten (bezeich](https://thumb-eu.123doks.com/thumbv2/1library_info/4565386.1599813/25.892.123.784.109.492/abbildung-pulverförmigen-materials-kristallinen-komplexes-vergleich-theoretisch-berechneten.webp)
![Tabelle 6 U–Cl-Abstände in der Kristallstruktur des [HDipp 2 Im] 2 [UCl 6 ]∙MeCN-Komplexes](https://thumb-eu.123doks.com/thumbv2/1library_info/4565386.1599813/31.892.109.789.783.1017/tabelle-cl-abstände-kristallstruktur-hdipp-ucl-mecn-komplexes.webp)
