RISIKOFAKTOREN FÜR DIE GRAFT-VERSUS HOST DISEASE DER LEBER
104
0
0
Volltext
(2)(3)
(4)
(5)
(6)
(7)
(8)
(9)(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
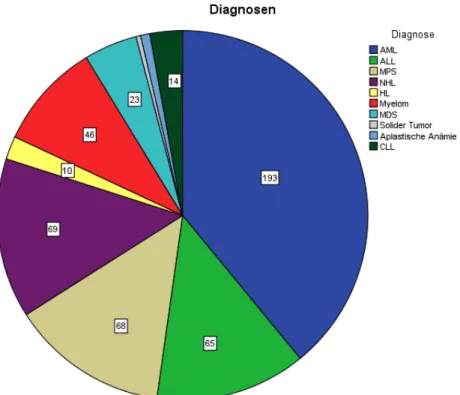
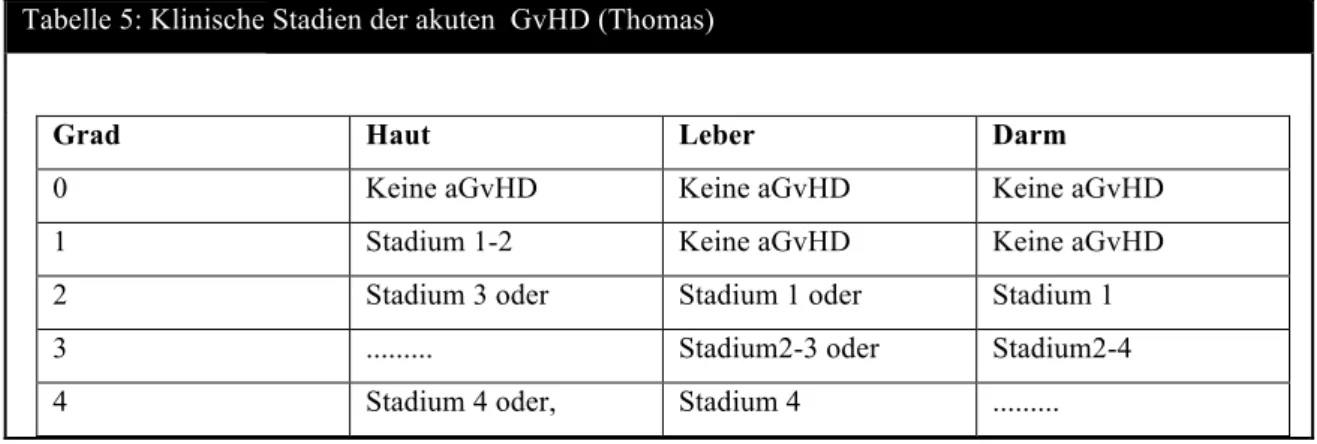
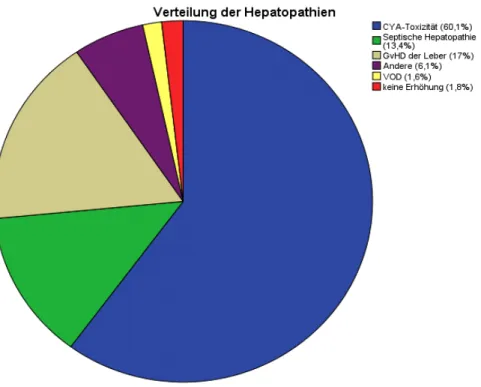
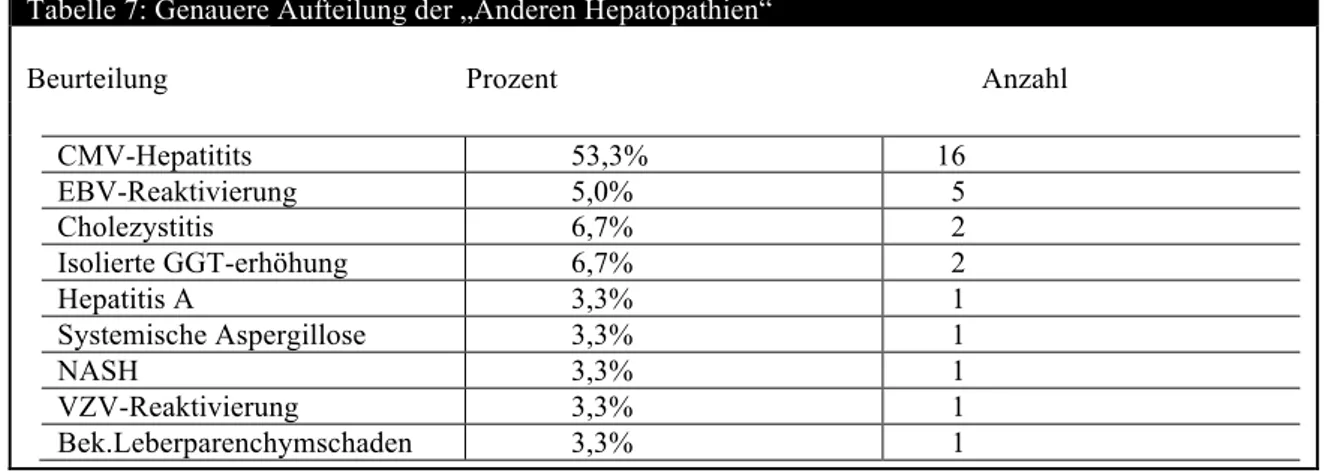
Abbildung


+7
ÄHNLICHE DOKUMENTE
Donor T cells deficient for miR-142 show impaired cell cycle progression and simultaneously reduced proliferation, which in turn leads to reduced GvHD severity and mortality
Kanfer, et al., Graft invariant natural killer T-cell dose predicts risk of acute graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Coman,
In our study, the selected genes encoding chemokines were all up-regulated in mice transferred with GC-resistant allogeneic T cells compared to mice transplanted with
Gut 30 Jahre nach der Conter- gan-Katastrophe wurde das Sedati- vum Thalidomid von amerikanischen Onkologen zur Behandlung der chronisch verlaufenden Graft-Ver-
Arnold, NIH-Defined Chronic Graft-Versus-Host Disease and Graft-Versus- Leukemia Effects in Patients Receiving Allogeneic Hematopoietic Cell Transplantation for Acute
DP/CD4SP ratios were in the majority of these mice not distinguishable from ratios in OT-II b →[d→C57BL/6] non-deleting controls (no thymic mOVA). Deficient elimination
The overall hypothesis underlying this thesis postulates that GVHD- mediated injury to the thymus, in particular to the thymic epithelial cell compartment, is a
„Nach der Transplantation ändert sich auch die Blutgruppe des Patienten“, so Zeiser, „ein Blutgruppen-Switch, weil alle Blutzellen neu gebildet werden.“ Dabei sind die
