Untersuchung der Struktur und Funktion von Sieben-Helix-Membranrezeptoren
142
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
Abbildung
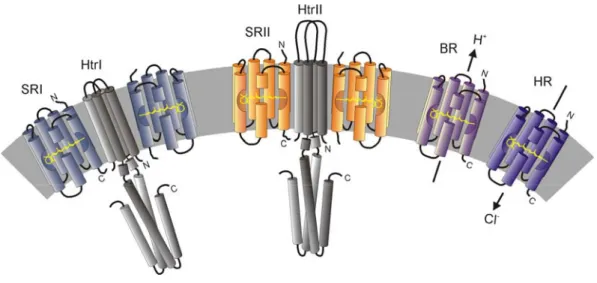

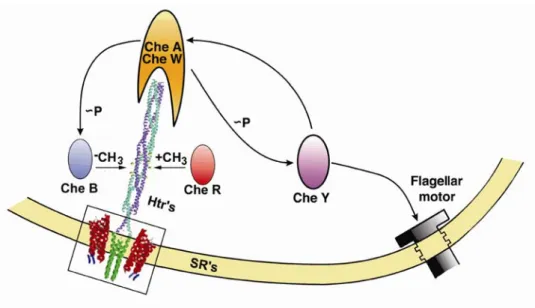
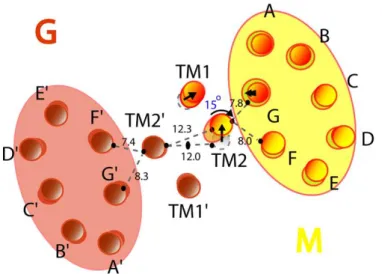
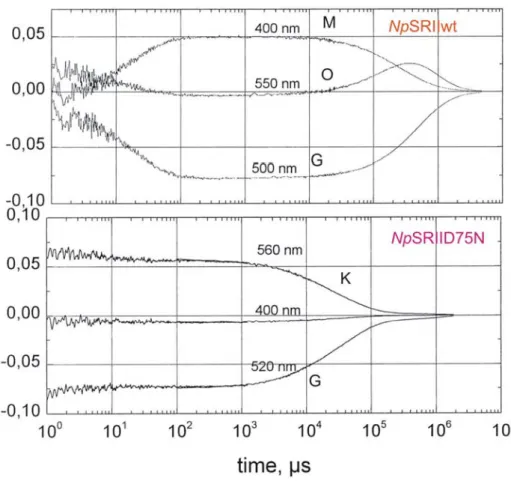
+7
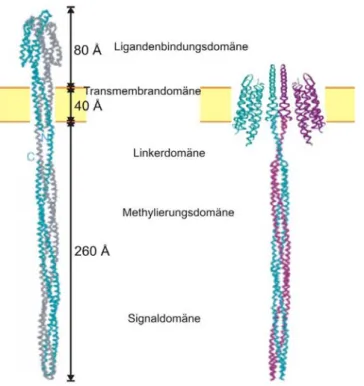
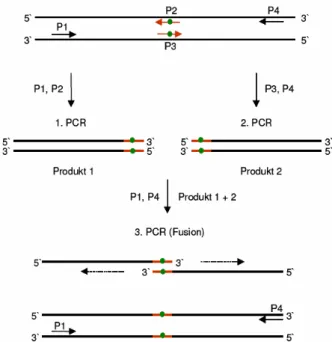
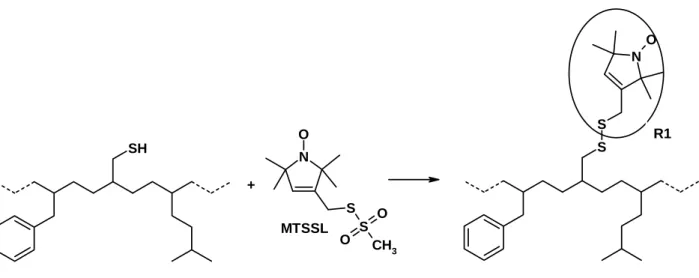
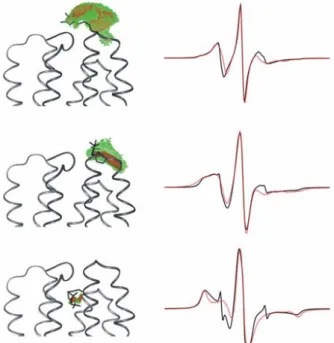
![Abb. 3.1.1: Kristallstruktur des Rezeptor-Transducer-Komplexes NpSRII-NpHtrII 157 [1H2S] mit ausgewählten Positionen für Spinmarkierung](https://thumb-eu.123doks.com/thumbv2/1library_info/3650158.1503270/55.892.241.666.420.828/kristallstruktur-rezeptor-transducer-komplexes-nphtrii-ausgewählten-positionen-spinmarkierung.webp)
ÄHNLICHE DOKUMENTE
Antikörper welche nur PF4/Heparin-Komplexe erkennen (aber nicht PF4 alleine). Dann haben wir die Kräfte gemessen, die entstehen, wenn die Spitze mit den daran gebundenen Antigenen mit
Mittels RT-PCR konnte auch eine ektopische Expression von CLEC3A in verschiedenen Geweben nachgewiesen werden.. Dies ließ sich auf Proteinebene jedoch
Allerdings wird das Referenzstrahl-Signal nicht durch die Atmo- sphär (inklusive Plexiglas-Platte) sondern durch eine externe Referenzquelle erzeugt. 0 Eine Gruppe der
Hinzu kommt, dass die wenigen Studien, in denen die Prävalenz sowohl subjektiver als auch objektiver Tagesschläfrigkeit erhoben wurde, die Ergebnisse der einzelnen Tests in
Pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial OS=Bos taurus GN=PDHA1 PE=2 SV=1 ODPA_BOVIN (+7) Pyruvate dehydrogenase E1 component subunit
Um diese aus Sicht des Steuerpflichtigen negativen steuerlichen Folgen der Über- tragung von Wirtschaftsgütern auszuschließen und um auch künftig die Betriebsaufspaltung als