Detektion photokatalytischer Aktivität aus der Einzelmolekülfluoreszenz organischer Farbstoffe Detektion photokatalytischer Aktivität aus der Einzelmolekülfluoreszenz organischer Farbstoffe Detektion photokatalytischer Aktivität aus der Einzelmolekülfluor
171
0
0
Volltext
(4)(5)
(6)(7)
(8)(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(32)
(33)
Abbildung
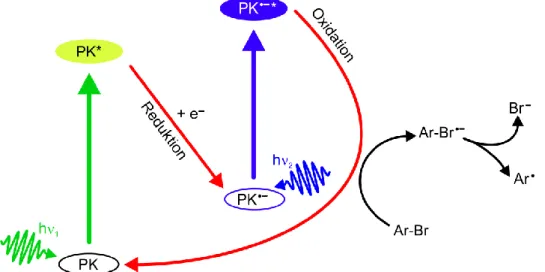
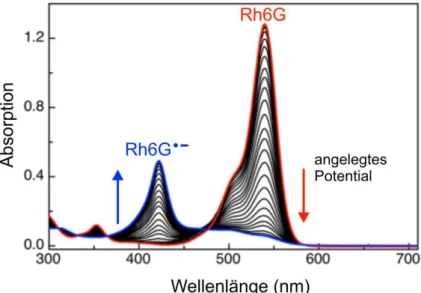
+7


ÄHNLICHE DOKUMENTE
Er ist mit einer Vierfach-Filterkombination ausgestattet und nutzt Vorfilter, Aktivkohle- und Schwebstoff- (HEPA) sowie UV-LED-Fotokatalyse- Filter, um das gesamte Spektrum
März 2011 in Bergisch Gladbach Lina Neunhäuserer
Die Teilnehmerinnen und Teilnehmer haben sich im Rahmen dieses Seminars aktiv mit der Zukunft ehrenamtlicher Tätigkeit im Bereich „Hilfe für Angehörige psychisch Erkrankter“
Proteinkinasen nehmen eine Schlüsselposition in den zellulären Signaltransduktionswegen ein. Sie können für die Entartungen von Zellen, die bei Krebs oder
Nach Ausschalten der Cy5 Fluoreszenz durch Anregung mit 633 nm (rot schraffierter Bereich), wurde für 2.5 Sekunden mit der blauen Laserwellenlänge belichtet. Das
Nicht nur der Verfasser der vorstehend erwähnten Broschüre «Die schweizerische Abrüstung», sondern auch die sozialistische Presse der Schweiz hat sich neuestens wieder den
bürgerliche Zeitung der deutschen Schweiz Hess es sich nicht nehmen, im Textteil ihrer Fremdenverkehrs-Beilage für die menschenfreundliche Wirtin des Freizeitheims im Tessin und
Die TNs stehen danach ein wie viele Gemüse und Früchte sie gerne haben und eine der Gärtnerinnen teilt dann die Kinder in Gruppen ein.. Jede Gruppe hat
