Research Collection
Doctoral Thesis
Mining the sequence space of antibody repertoires to predict and design antigen-specific antibodies
Author(s):
Friedensohn, Simon Publication Date:
2020
Permanent Link:
https://doi.org/10.3929/ethz-b-000454740
Rights / License:
In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use.
DISS. ETH NO. 26686
Mining the sequence space of antibody repertoires to predict and design antigen-specific antibodies
A thesis submitted to attain the degree of DOCTOR OF SCIENCES of ETH ZURICH
(Dr. sc. ETH Zurich)
Presented by SIMON FRIEDENSOHN M.Sc. in Biotechnology, ETH Zurich
Born on 10.04.1990
citizen of the Federal Republic of Germany
accepted on the recommendation of
Prof. Sai Reddy, examiner Prof. Bruno Correia, co-examiner
Prof. David Gfeller, co-examiner
2020
“It is very easy to overestimate the importance of our own achievements in comparison to what we owe others.”
- Dietrich Bonhoeffer
Zusammenfassung
Das adaptive Immunsystem von Säugetieren ist in der Lage, spezifische molekulare Strukturen auf körperfremden Krankheitserregern zu identifizieren. Die Spezifität für diese Epitope wird durch eine Gruppe von Rezeptoren erreicht, die zur Immunglobulin-Superfamilie gehören: B-Zell-Rezeptoren (BCR), ihre sezernierte Version (Antikörper) und T-Zell-Rezeptoren (TCR). Jeder dieser Rezeptoren trägt hochvariable Regionen, die die Antigenerkennung erleichtern und die während der Entwicklung der Vorläuferzellen erzeugt werden (und daher als einzigartige Klone oder klonale Linien angesehen werden). Die aktuelle Schätzung für die theoretische Vielfalt einzigartiger naiver BCR-Sequenzen liegt bei 5x10
13klonalen Umlagerungen für den Menschen und mindestens 10
12für die Maus. Die vielfältige Population von BCRs, Antikörpern oder TCRs in einem bestimmten Individuum wird als Immunrepertoire bezeichnet.
Zur Sequenzierung des Immunrepertoires (AIRR-Seq, Ig-Seq) wird die
Tiefensequenzierung verwendet, die auf diese große Vielfalt in den verschiedenen
immunologischen Kompartimenten und Immunzell-Subsets zugreifen und sie
analysieren kann. Diese enorme Informationsfülle hat zu neuen Erkenntnissen in
Bereichen wie zum Beispiel des Antikörperentwicklung, der Immundiagnostik, des
Impfstoffdesigns und der Immunologie geführt. In Kapitel 1 dieser Dissertation
erörtere ich die aktuellen Trends in der Sequenzierung des Immunrepertoires und die
Bemühungen zur Verbesserung der bestehenden Protokolle in Bezug auf die
Exaktheit und Qualität der Sequenzierdaten. Ich hebe einige der größten
Herausforderungen in diesem Bereich hervor, wie z.B. die mangelnde Genauigkeit
und die Sequenzierung von gepaarten variablen Regionen (z.B. der schweren und
leichten Kette). Da beispielsweise das Erstellen der Sequenzierbibliothek und die
Plattformen für die Tiefensequenzierung Fehler und Verzerrungen verursachen
können, kann dies die immunologische Interpretation beeinträchtigen. Dies ist
besonders herausfordernd im Zusammenhang mit B-Zellen, die sich einer
somatischen Hypermutation unterziehen, einem natürlichen Prozess, der Mutationen
in die variablen Antikörperdomänen einführt.
Zusammenfassung
In Kapitel 2 beschreibe ich eine experimentelle und rechnergestützte Methode, die wir auf der Grundlage synthetischer Standards und molekularer Strichkodierung („molecular barcoding“) entwickelt haben und die implementiert wurde, um eine hochpräzise Sequenzierung des Antikörperrepertoires zu erreichen. Wir zeigen, dass dieses konzeptionell einfache Verfahren es uns ermöglicht, die Fehlerraten über die gesamte Sequenzierungsregion hinweg deutlich zu reduzieren. Anhand menschlicher B-Zell-Proben zeigen wir, dass diese Technik die Messungen von Antikörper- repertoires hinsichtlich verschiedener Parameter verbessern kann.
Obwohl es nun möglich ist, qualitativ hochwertige Ig-Seq-Datensätze zu erstellen, ist die Verknüpfung von Sequenz und Antigen-Spezifität eine immens anspruchsvolle Aufgabe. In Kapitel 3 gebe ich eine Einführung in das Konzept der Modellierung des großen Sequenzraums von Immunrepertoires, mit dem Ziel deterministische Sequenzmotive zu extrahieren, die mit der Antigenexposition und - spezifität korrelieren. Ich bespreche verschiedene Klassen von statistischen und maschinellen Lernalgorithmen, die zur Modellierung der Sequenzerzeugung verwendet werden können.
In Kapitel 4 entwickle ich einen neuartigen Ansatz zur Identifizierung antigenspezifischer Sequenzmuster in Antikörperrepertoires, der auf tiefen generativen Modellen beruht. Zur Modellierung des zugrundeliegenden Prozesses der BCR-Generierung wurden Variationsautokodierer (VAE) verwendet, bei denen angenommen wurde, dass die Datengenerierung im Latenzraum („latent space“) einem Gauss-Mischverteilungsmodell (GMM) folgt. Dies lieferte sowohl eine latente Einbettung („latent embedding“) als auch Gruppenkennzeichnungen, die ähnliche Sequenzen zusammenfassen, was eine Vielzahl konvergenter, antigenassoziierter Sequenzmuster ergab. Diese antigenassoziierten Sequenzmuster waren prädiktiv für die immunologische Geschichte und stellen antigenbindende Antikörper dar.
Schließlich zeige ich, wie diese Sequenzmuster genutzt werden können, um weitere
antigenspezifische Antikörper
in silico zu generieren, die abschliessend auchexperimentell auf ihre Antigenspezifität überprüft wurden.
Abstract
Abstract
The mammalian adaptive immune system is able to identify specific molecular structures on foreign pathogens. Specificity to these epitopes is achieved through a group of receptors belonging to the immunoglobulin superfamily: B cell receptors (BCR), their secreted version (Antibodies) and T cell receptors (TCR). Each of these receptors carries highly variable regions, which facilitate antigen recognition and which are generated during progenitor cell development (and thus are thought to be unique clones or clonal lineages). The current estimate for the theoretical diversity of unique naïve BCR sequences is around 5x10
13clonal combinations for humans and at least 10
12for mice. The diverse population of BCRs, antibodies or TCRs in a given individual is referred to as the immune repertoire.
Immune repertoire sequencing (AIRR-Seq, Ig-Seq) utilizes deep sequencing to access and analyze this vast diversity in different immunological compartments and immune cell subsets. This massive wealth of information has generated novel insights in the fields of antibody engineering, immunodiagnostics, vaccine design, as well as basic immunology. In Chapter 1 of this thesis, I review the current trends in immune repertoire sequencing and the efforts taken to improve existing protocols in relation to accuracy and quality of the sequencing data. I highlight several of the most major challenges in the field, such as obtaining paired variable region (e.g., variable heavy and variable light) sequencing and a lack of accuracy. For example, since sequencing library preparation and platforms for deep sequencing can introduce errors and biases, it can compromise immunological interpretations. This is especially confounding in the context of B cells that undergo somatic hypermutation, a natural process that introduces mutations in antibody variable regions.
In Chapter 2, I describe an experimental and computational method we have
developed based on synthetic standards and molecular barcoding, which has been
implemented to achieve highly accurate antibody repertoire sequencing. We show
how this conceptually simple procedure allows us to significantly reduce error rates
across the whole sequencing region. By applying this technique to human B cell
Abstract
samples, we demonstrate that it can improve the measurements of antibody repertoires across various dimensions.
Although it is now possible to produce high quality Ig-Seq datasets, linking sequence to antigen-specificity is an immensely challenging task. In Chapter 3, I provide an introduction to the concept of modeling the large sequence space of immune repertoires in order to extract deterministic sequence motifs that correlate with antigen exposure and specificity. I review various classes of statistical and machine learning algorithms that can be used to model sequence generation.
In chapter 4 I develop a novel approach to identify antigen-specific sequence patterns in antibody repertoires based on generative deep models. To model the underlying process of BCR generation, variational autoencoders (VAE)s were used, where it was assumed that data generation follows a Gaussian mixture model (GMM) in latent space. This provided both a latent embedding and also cluster labels that group similar sequences together, which revealed a multitude of convergent, antigen- associated sequence patterns. These antigen-associated sequence patterns were predictive of immunological history and represent antigen-binding antibodies.
Finally, I demonstrate how these sequence patterns can be used to generate further
antigen-specific antibodies in silico, that are experimentally verified to retain antigen-
specificity.
Table of Contents
Table of Contents
Zusammenfassung ... iii
Abstract ... v
Table of Contents ... 1
List of Figures ... 3
List of Tables ... 4
Thesis Objectives ... 6
Chapter 1 General introduction ... 8
Generation of immune receptor repertoire diversity ... 8
The rapid rise of high-throughput immune repertoire sequencing ... 8
Next generation sequencing platforms used for immune repertoire sequencing ... 10
Improving accuracy in immune repertoire data ... 11
High-throughput pairing of variable regions ... 15
Concluding remarks and future directions ... 20
Chapter 2 ... 22
Abstract ... 23
Introduction ... 23
Results ... 25
Design of a comprehensive set of human synthetic standards ... 25
Human Ig-Seq library preparation using the MAF protocol ... 27
Combining standards with MAF to correct errors and bias in Ig-Seq ... 29
Impact of MAF error correction on human B cell repertoires ... 31
Clonal diversity measurements of human B cell repertoires after error and bias correction ... 34
Divergent features of CD27-IgM+ and CD27+IgG+ repertoires ... 37
Discussion ... 39
Methods ... 42
Chapter 3 ... 47
Protein sequence space and machine learning ... 47
Unsupervised machine learning in antibody repertoires ... 47
Modelling protein sequence space ... 48
Variational autoencoder with a mixture of gaussians prior ... 50
Table of Contents
Chapter 4 ... 53
Abstract ... 54
Results ... 54
Antigen-dependent sequence convergence in antibody repertoires ... 55
Convergent antibody sequences are antigen specific ... 59
Generative modelling of antibody sequences ... 62
Discussion ... 63
Methods ... 64
Chapter 5 General discussion ... 72
Chapter 6 Appendices ... 77
Supplementary Information – Chapter 2 ... 77
Supplementary Information – Chapter 3 ... 85
Chapter 7 References ... 98
Acknowledgements ... 110
Curriculum Vitae ... 111
List of Figures
List of Figures
Figure 1-1 Overview of Immune Repertoire Pairing and Sequencing Methods ... 16
Figure 1-2 Single-cell sequencing and immune receptors ... 20
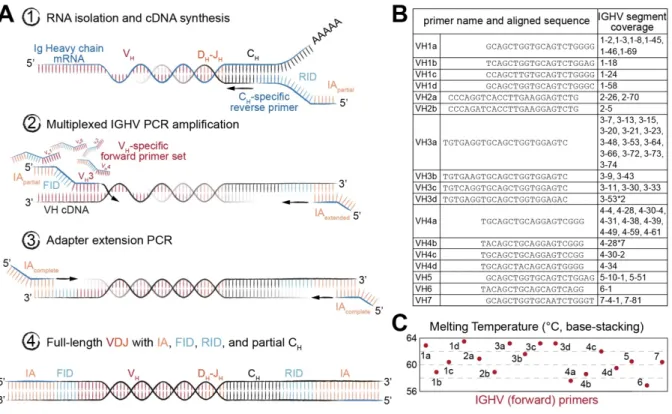
Figure 2-1 A comprehensive set of human synthetic spike-in standards for Ig-Seq ... 26
Figure 2-2 Library preparation of immunoglobulin (Ig) heavy chain genes for high-throughput sequencing (Ig-Seq) using molecular amplification fingerprinting (MAF) ... 28
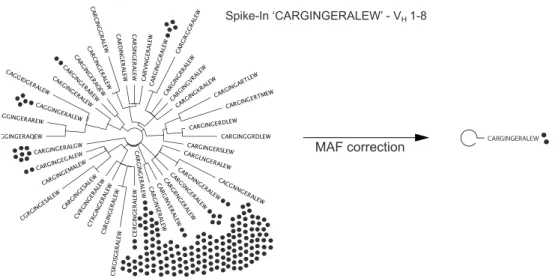
Figure 2-3 Phylogenetic tree visualizing CDRH3 variants ... 30
Figure 2-4 Synthetic standards used to validate performance of error and bias correction of Ig-Seq data by MAF ... 31
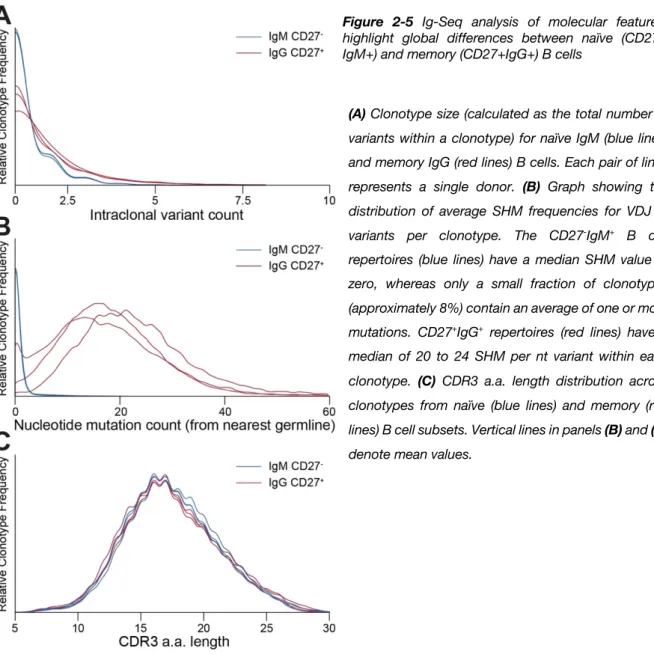
Figure 2-5 Ig-Seq analysis of molecular features highlight global differences between naïve (CD27-IgM+) and memory (CD27+IgG+) B cells ... 32
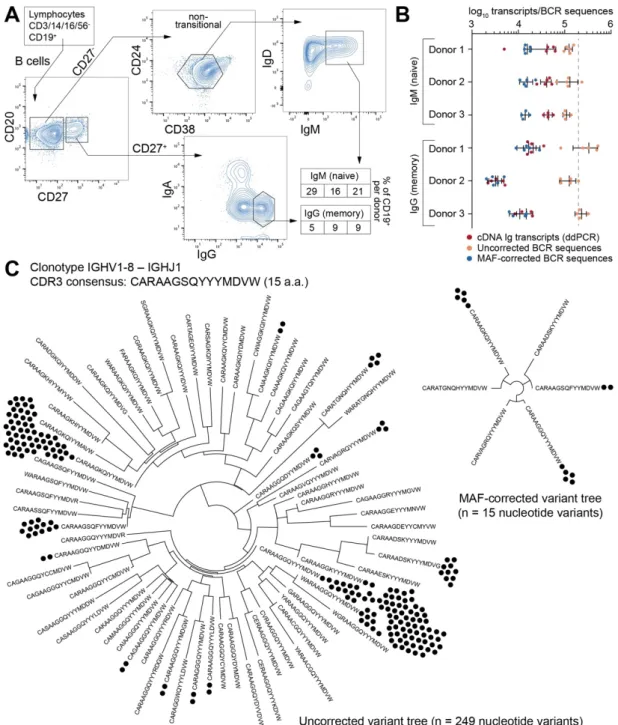
Figure 2-6 Ig-Seq analysis of human naïve (CD27-IgM+) and memory (CD27+IgG+) B cells ... 33
Figure 2-7 Clonotype diversity analysis across cellular replicates of naïve (CD27-IgM+) and memory (CD27+IgG+) B cells ... 36
Figure 2-8 Genetic features of naïve and IgG memory BCR repertoires ... 38
Figure 4-1 Schematic overview of the study ... 55
Figure 4-2 Identification and characterization of antigen-associated sequences ... 57
Figure 4-3 Latent space captures sequence features ... 58
Figure 4-4 Hybridoma library workflow ... 59
Figure 4-5 Antigen specific sequences across all repertoires ... 61
Figure 4-6 Latent antibody space and sequence generation ... 63
Supplemental Figures
Supplemental Figure 1 Sequencing Bias ... 77Supplemental Figure 2 Clonotype definition stringency does not create analysis bias comparing ... 78
Supplemental Figure 3 Clonotype estimation ... 79
List of Tables
Supplemental Figure 4 Linear discriminant analysis distinguishes CD27+ IgG repertoires
and CD27- IgM repertoires based on their V-Gene ... 80
Supplemental Figure 5 Clonal composition ... 81
Supplemental Figure 6 Accuracy ... 86
Supplemental Figure 7 Sequencing results ... 87
Supplemental Figure 8 GMM-VAE architecture ... 88
Supplemental Figure 9 Additional latent space visualizations ... 89
Supplemental Figure 10 Cluster-based prediction; Cross-validation results ... 89
Supplemental Figure 11 Sequence based prediction; Cross-validation results ... 90
Supplemental Figure 12 Cross-validation procedure ... 90
Supplemental Figure 13 Exemplary sequence cluster ... 91
Supplemental Figure 14 V-Gene usage ... 91
Supplemental Figure 15 Alignment of antigen specific heavy chain sequences ... 91
Supplemental Figure 16 Flow cytometry analysis of hybridoma libraries ... 92
Supplemental Figure 17 ELISA profiles of antigen-specific hybridoma clones ... 93
Supplemental Figure 18 Alignment of natural variants ... 93
Supplemental Figure 19 ssODN library workflow ... 93
Supplemental Figure 20 Sampling results ... 94
Supplemental Figure 21 Library results ... 94
List of Tables
Table 1-1 Methods for correcting immune repertoire sequencing data ... 12Table 1-2 A brief overview of single-cell sequencing technologies ... 19
Table 4-1 Screened convergent variants ... 60
Supplemental Tables
Supplemental Table 1 Molecular characteristics of 85 synthetic human IgH gene standards ... 82List of Tables
Supplemental Table 2 Sequencing results I ... 83
Supplemental Table 3 Sequencing results II ... 83
Supplemental Table 4 Primers and probes used in this study ... 84
Supplemental Table 5 Immunization scheme ... 85
Supplemental Table 6 Binder sequences – Ovalbumin ... 85
Supplemental Table 7 Surrogate light chain sequences for OVA1, OVA5 ... 86
Supplemental Table 8 Binder sequences - RSV-F ... 86
Supplemental Table 9 Surrogate light chain for RSV1, 2 and 3 ... 86
Supplemental Table 10 Overview over sampled sequences ... 94
Supplemental Table 11 ... 95
Supplemental Table 12 ... 96
Supplemental Table 13 Convergent clones identified via various clustering approaches .. 97
Thesis Objectives
Thesis Objectives
This thesis aims to address the following research questions:
Improving Ig-Seq (Chapter 1 & 2)
In most cases, preparing high-throughput sequencing libraries involves one or several PCR steps. Especially in Ig-Seq, one of the most commonly used approaches is a two-step PCR method during which the flowcell adapters for Illumina sequencing are added [1]. However, PCR is a noisy process which adds artificial mutations to the original sequence. In classical applications such as standard RNA-Seq, these errors often do not matter that much since one is still able to align erroneous transcripts back to their reference. However, in Ig-Seq knowing the exact sequence is of paramount importance, for example when we want to express the actual antibody sequence to evaluate its binding behavior. Other cases such as the construction of correct clonal lineage trees are also very challenging when working with uncorrected data. Furthermore, many Ig-Seq protocols use a multiplexed primer set to amplify the wide variety of natural occurring antibody sequences. However, these primer sets often do add considerable sequencing bias to the assay, which causes problems when one is interested in the correct rankings or frequencies of B cell clones in a population. One proposed solution to this problem has been first reported for virus sequencing [2]. Here, a degenerate stretch of nucleotides is added to the reverse transcription primer in order to tag individual cDNA molecules. This tag can be used to count cDNA/RNA molecules directly and thus correct the observed sequencing bias. Additionally, when we oversample on these unique molecular identifiers (after PCR), we will be able to correct potential PCR and sequencing errors by grouping sequences by their UMI and correcting diverging positions by a majority voting scheme. One objective of this thesis is to develop the experimental conditions to incorporate these UMIs into human Ig-Seq and to develop the appropriate computational pipeline to analyze this data and make it accessible to other people working in this field.
Modeling antibody sequences (Chapter 3 & 4)
It is likely to assume that only a smaller (although in practice still huge) subset of the theoretically possible set of B cell and T cell receptor sequences can be generated due to physical and chemical constraints. Furthermore, receptor-antigen interaction also constrains the path along which an antibody can evolve. Modelling the generative process of this functional subset of antibodies can be used to simulate novel variants of already existing
Thesis Objectives
antibodies in a principled manner. Here we evaluate whether a model based on a variational autoencoder possess the capability to learn these underlying rules from the data itself and whether this model can be used to make predictions about unseen sequences. In the long term such a tool will be valuable when making principled decision about the rational engineering of antibodies.
Identifying convergent antibodies (Chapter 4)
Several recent studies have reported the occurrence of similar or identical antibody and TCR sequences across human patients and model organisms with comparable disease or immunization history [3-5]. The goal of this part of the thesis is to identify methods that robustly identify immunological status based on observed antibody sequences in a repertoire. As a proof of concept, we plan to sequence the B cell repertoires (Bone Marrow) of several, large cohorts of immunized mice, that have been immunized with three model antigens (Hen Egg Lysozyme, Ovalbumin, Blue Carrier Protein) and one pharmaceutically relevant antigen (Respiratory Syncytial Virus Fusion (RSVF) protein). Another avenue of research is whether we could reduce the feature size of our datasets by finding a better representation of our sequences. For example, it might be plausible to assume that a certain degree of sequence similarity is enough to confer protection towards a specific antigen. Thus, it might be more valuable and powerful to identify predictive clonotypes or clonal lineages instead of specific antibody clones / sequences or find a set of different biophysical descriptors by which antibody sequences could be grouped.
Expressing predicted and modeled antibody sequences (Chapter 4)
Lastly, we want to express some of the antibodies that we previously identified or modeled and then characterize them for their binding behavior with simple ELISAs.
Thus, a summary of my thesis objectives is the following: We want to develop tools to acquire better biological data from our Ig-Seq experiments. Afterwards we want to utilize the obtained data to discover and model novel antibodies, which finally we want to express and experimentally validate, thereby presenting a principled and novel way of monoclonal antibody discovery and engineering. We conclude this dissertation with a final discussion and outlook in Chapter 5.
General introduction
General introduction
This introduction is an author produced adaptation and expansion of a peer reviewed review article published in Trends in Biotechnology in 2017. [Simon Friedensohn, Tarik A. Khan, Sai T. Reddy] doi: https://doi.org/10.1016/j.tibtech.2016.09.010
Generation of immune receptor repertoire diversity
Lymphocyte T-cell receptors (TCR), B-cell receptors (BCR) and immunoglobulins (Ig)/antibodies (secreted form of BCRs) encode a vast functional sequence space. This diversity is mainly confined to receptor variable regions, which facilitate the recognition and binding of the myriad of pathogens and antigens encountered by a host. Besides carrying these variable regions, the BCR and TCR are structurally related. The TCR is made up of two protein sub-units linked by disulfide bonds (the α and the b chain in most cases, although some TCRs are comprised of the so called γ/δ chains), both of which carry the aforementioned variable region as well as a constant region. Likewise, antibodies (both, the membrane bound version as well as the secreted form) consists of two chains, the light and the heavy chain. However, an antibody is formed by two identical heavy and two identical light chains – again linked by disulfide bonds – giving rise to its final “Y-shaped” form.
Formation of variable regions is a result of the somatic recombination, called V(D)J recombination, of distinct germline gene elements – variable- (V-), diversity (D-) and (J-) joining-segments. For each segment multiple different variants exists in the respective genomic loci. The variable region of heavy and b chain is formed by recombining only a V, D and J segment, whereas the light and α chain only utilize a V and J segment. In humans, these recombination can generate a theoretical diversity of at least 5x1013 naïve BCRs and 1018 α:b TCRs [6]. However, only a subset of this diversity is expected to be physiologically present in the actual repertoire of an individual [7, 8]. In addition to V(D)J recombination, B- cells also undergo a process of secondary diversification through somatic hypermutation, a highly regulated enzymatic process that introduces additional mutations in variable regions in order to increase the affinity of BCRs to antigen targets. In the past, empirically measuring the enormous diversity of lymphocyte repertoires was relegated to qualitative approaches such as complementarity determining region 3 (CDR3) spectratyping or Sanger sequencing of small subsets of clones (i.e., ~10-100).
The rapid rise of high-throughput immune repertoire sequencing
The rapid rise of high-throughput immune repertoire sequencing (see Glossary) has led to unprecedented quantitative insight into lymphocyte diversity and adaptive immunity, leading
General introduction
to a new era of systems immunology. Here, we will refer to this paradigm for T cells and B cells as TCR-Seq and Ig-Seq, respectively [9, 10]. In one of the first examples using TCR- Seq, Robins et al. quantitatively measured human TCR diversity [11], while subsequent studies using revealed the clonal overlap of repertoires shared between individuals was higher than anticipated based on theoretical calculations [12-14]. These seminal studies not only provided quantitative and in some cases unexpected answers to long-standing questions in basic lymphocyte immunobiology, but also established the basic experimental and computational methods needed for high-throughput repertoire sequencing. Moreover, they laid the foundation for a breadth of follow-up studies including the implementation of TCR-Seq and Ig-Seq for applied research.
In clinical settings, TCR-Seq has been performed on patients with acute T-lymphoblastic leukemia before and after treatment, revealing the receptor sequence and frequency of potential neoplastic lymphoblasts [15]. This approach was further developed as a clinical assay for diagnosis of minimal residual disease (MRD), which describes the subset of malignant lymphocytes that remain during and after the course of treatment. TCR-Seq provided equal or higher sensitivity to conventional tests (flow cytometry- or PCR-based) [11, 15-17]. For biotechnology applications, Ig-Seq has become a valuable tool for monoclonal antibody (mAb) discovery and engineering. For example, Ig-Seq was performed on plasma cells of immunized mice in order to identify antigen-specific mAbs based on sequencing information alone, thus bypassing cost- and time-intensive screening procedures [18]. Ig- Seq on human B cells has been used to identify variants of broadly neutralizing antibodies against HIV-1 [19, 20]. Another innovation has been to use shotgun proteomics workflows to obtain peptide mass spectra data of affinity-purified, polyclonal antibody proteins, whereby the sequences of these peptides are identified using a reference database generated by Ig- Seq [21-23]. This approach enables a serological and molecular analysis of the antibody proteome, revealing information like the presence of post-translational modifications, which is not possible with Ig-Seq alone[24]. In vaccine profiling, preliminary studies have shown that Ig-Seq can be used to determine the dynamics and developmental lineage of B cells following seasonal influenza vaccination [25, 26]. The studies described above demonstrate the gaps within applied immunology that are being overcome using immune repertoire sequencing.
While rapid progress in immune repertoire sequencing has already led to a number of important advances, there are still several challenges that must be overcome before these techniques fully realize their potential. Here, we give a short introduction into recent innovations in TCR-Seq and Ig-Seq which aim to solve these main challenges. The first
General introduction
problem is the presence of errors and bias in sequencing datasets, which are introduced through library preparation methods (e.g., Multiplex-PCR, 5’ RACE) and NGS platforms themselves [27, 28]. While artifacts in datasets are a concern for any NGS-based application, they are particularly troublesome for immune repertoires given that their diversity and distribution directly impact immunological interpretations. A second limitation arises from the fact that many basic repertoire sequencing protocols rely on using bulk cell populations as input, which does not preserve information about variable region pairing. Since variable region pairing is often essential for determining antigen specificity, this also limits the functional conclusions that can be made with TCR-Seq and Ig-Seq. In order to achieve true and comprehensive immune repertoire knowledge, the field must adopt error/bias correction methods, variable region pairing, and potentially further relevant information (e.g.
(epi)genomic and transcriptomic data).
Next generation sequencing platforms used for immune repertoire sequencing
Several NGS systems have been used for immune repertoire sequencing; these include instruments from IonTorrent [29], Pacific Biosciences [30, 31], Roche/454 Life Sciences [11]
and Illumina [32-34]. Similar to the fields of genomics and transcriptomics, Illumina-based sequencing has become the most widely used technology in immune repertoire sequencing [10]. Each of these NGS platforms vary in the amount and type of errors that are added during base calling. For example, while Illumina’s MiSeq instrument is reported to have a relatively low error rate, a recent study indicated that the percentage of error-free reads of a 150 bp, paired-end sequencing run was close to only 80% [35]. The number of erroneous read is likely to be even greater for immune repertoire datasets because longer sequencing runs are necessary in order to sequence full-length variable regions (~350-420 bp). Although other instruments show higher error-rates than Illumina’s MiSeq, their usage might be preferable under certain circumstances. Longer read lengths (>1 kb) generated by Pacific Bioscience technology is useful for receptor pairing protocols, which physically link variable regions together [31, 36], while the fast sequencing turnover achieved with IonTorrent’s semiconductor technology is potentially interesting for clinical applications [35]. The recent improvements in nanopore sequencing can potentially obtain ultra-long reads (> 100 kb), which may facilitate researchers to screen the complete naïve or rearranged immune loci more efficiently [37]. This development could lead to the discovery of novel immune receptor diversification mechanisms, such as recently reported chromosomal integrations into variable regions [38].
General introduction
Improving accuracy in immune repertoire data
Sequencing errors and biases are generated in a number of ways [39]. In some cases, they can be mitigated by good laboratory practices, such as preventing degradation or contamination of the source material. However, there are inherent problems due to the underlying dependence of molecular biology-based techniques for NGS. These problems arise because the amount of starting material (genomic DNA or total RNA) is often limited and of low purity, thus requiring utilization of reverse transcriptases and/or DNA polymerases that are unable to perfectly transcribe or amplify the original material. While the use of high- fidelity enzymes (such as newly engineered proofreading reverse transcriptases [40]) and careful experimental design can partially reduce error rates, the rate of nucleotide misincorporation will still occur and propagate due to the exponential nature of PCR [41, 42]
and further errors can be added through the sequencing process itself. Additionally, in many cases, multiplex-PCR is conducted, where degenerate primer sets target either the beginning of the variable region or the leader region (further upstream of each V-segment) [1]. This introduces considerable bias towards preferentially amplified sequences [43]. As a means to circumvent these steps, several protocols have been adapted for TCR-Seq and Ig-Seq based on template-switching reactions that allow a single forward primer to be used (e.g., 5’RACE) [32, 44-46]. However, a recent study on amplification efficiency of TCR variable regions demonstrated that even when using a single primer and 5’ RACE, there was still preferential and biased amplification [27], most likely due to the nature of the template switching reaction.
The accumulation of errors in repertoire sequencing leads to an artificial inflation of observed clones or variable regions. Prudent filtering of sequencing reads based on quality-scores is a viable strategy to remove some false-positive reads and improve diversity estimates [47].
If paired-end sequencing data is generated (Illumina), merging both reads based on their overlap increases the quality of the overlapping region [48], which usually occurs in the clonal-identifying CDR3 of variable regions. The removal of scarce clones, which only have few reads mapping to them, has been suggested as a generic strategy to remove falsely identified sequences [12]. Besides these general guidelines, several more sophisticated methods to correct for substitution errors and to a lesser extent also for amplification bias have been applied to TCR-Seq and Ig-Seq (see Table 1-1) (for more details please refer to a recent review, Greiff et al. [49]).
General introduction
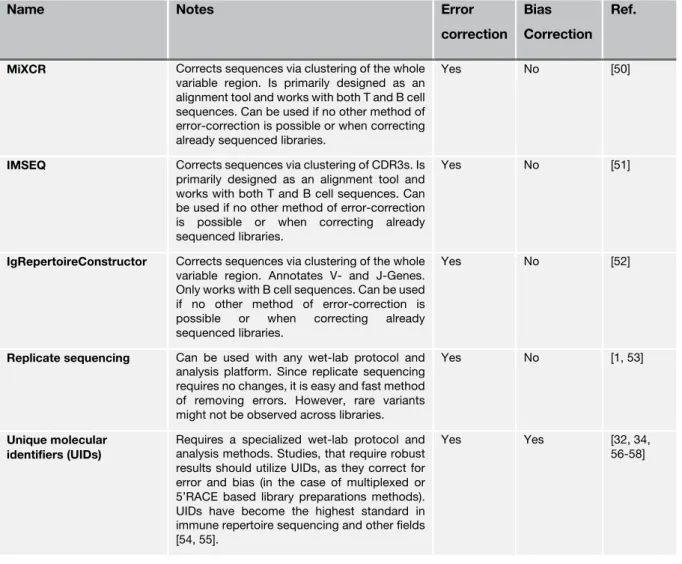
Table 1-1 Methods for correcting immune repertoire sequencing data
Name Notes Error
correction Bias Correction
Ref.
MiXCR Corrects sequences via clustering of the whole variable region. Is primarily designed as an alignment tool and works with both T and B cell sequences. Can be used if no other method of error-correction is possible or when correcting already sequenced libraries.
Yes No [50]
IMSEQ Corrects sequences via clustering of CDR3s. Is primarily designed as an alignment tool and works with both T and B cell sequences. Can be used if no other method of error-correction is possible or when correcting already sequenced libraries.
Yes No [51]
IgRepertoireConstructor Corrects sequences via clustering of the whole variable region. Annotates V- and J-Genes.
Only works with B cell sequences. Can be used if no other method of error-correction is possible or when correcting already sequenced libraries.
Yes No [52]
Replicate sequencing Can be used with any wet-lab protocol and analysis platform. Since replicate sequencing requires no changes, it is easy and fast method of removing errors. However, rare variants might not be observed across libraries.
Yes No [1, 53]
Unique molecular identifiers (UIDs)
Requires a specialized wet-lab protocol and analysis methods. Studies, that require robust results should utilize UIDs, as they correct for error and bias (in the case of multiplexed or 5’RACE based library preparations methods).
UIDs have become the highest standard in immune repertoire sequencing and other fields [54, 55].
Yes Yes [32, 34,
56-58]
Error correction via replicate sequencing
One approach for error correction relies on using technical replicates (e.g. sequencing libraries independently prepared from the same source material); after which, sequencing results obtained from each replicate are then validated against each other [39]. Greiff et al.
used this procedure to establish a reliability detection cutoff for excluding erroneous reads in Ig-Seq data [1, 53]. Ig-Seq reads were ranked by abundance and compared across replicate datasets, only the set of clones (defined by their CDR3) that were present in the respective replicate datasets at a specified fraction (e.g., 95%) were retained. This filtering procedure reduced the number of uniquely detected clones by 10- to 40-fold while still retaining roughly 95% of all sequencing reads. Similar numbers have been reported by Warren et al. [12]. However, when applying replicate filtering for the entire variable (VDJ) region, considerably fewer reads were retained (around 50%), highlighting that errors accumulate across the whole sequencing amplicon. These errors are especially problematic in Ig-Seq because they cannot be distinguished from naturally-occurring and biologically
General introduction
relevant mutations introduced by somatic hypermutation. While replicate sequencing helps to identify reliable full-length variable regions, the loss of up to 50% of the data and the need to at least double the sequencing depth via replicates increases the costs of this approach.
Rare sequences that are not shared across replicates will also not be found. Furthermore, the existence of reproducible hotspot PCR and sequencing errors is now well known, suggesting certain erroneous sequences derived from highly abundant clones will be present across replicates [32, 35, 59] .
Error correction through sequence clustering
The clustering of highly similar sequences has been another approach to error correction, which has the advantage of being able to use nearly all the reads generated by an NGS run.
In immune repertoire sequencing, clustering is often performed by grouping together sequences based on their CDR3 (or even complete V(D)J sequence) using a distance metric like the Hamming or Levensthein distance [11, 14, 51, 52]. In order to avoid grouping together highly homologous, yet distinct sequences, some algorithms also separate CDR3s into their constituent V-, D-, and J-gene segments. Clones are then not only separated by one distance metric across the whole CDR3, but by several metrics, one for each of the junctional segments [29]. The actual sequence or clone representing each cluster is generally identified by building the consensus sequence of all sequences in a given cluster. While effective for TCR-Seq, clustering and correcting errors solely based on CDR3 is less effective for Ig-Seq datasets generated from antigen-experienced B cells. This is because somatic hypermutations lead to the generation of multiple clones that share the same CDR3 sequence but differ at various other mutational hotspots across the rest of their V(D)J region. Thus, correcting sequences based on CDR3 alone will result in over-correction by removing unique somatic variants and reducing diversity. MiXCR, a recently developed immune repertoire annotation software, performs heuristic error-correction of variable regions by multilayer clustering of highly similar sequences [50], a method which tries to model the underlying error processes experienced by a set of immune repertoire sequences. If the user-specific criteria are chosen wisely and the underlying nature of the dataset allows for it, MiXCR’s algorithm is able to distinguish between true somatic variants and erroneous variants in Ig-Seq.
However, identifying the correct parameters is challenging and likely varies depending on the input material. It may be valuable for future studies to compare the analysis results from MiXCR to other modes of error-correction such as replicate sequencing or advanced experimental-bioinformatic workflows (see below).
General introduction
Error and bias correction with unique molecular identifiers
One of the most powerful error-correction strategies developed is based on unique molecular identifiers (UIDs) [2, 55]. Unlike the above in silico methods, UID-based correction requires additional experimental library preparation steps, but has the distinct advantage of offering capabilities to correct both errors and amplification bias. All UID methods rely on the single- molecule tagging of RNA or DNA molecules with a degenerate nucleotide sequence (e.g., NNNNN), which forms the basis of the UID. For immune repertoire sequencing, a common approach is to add these degenerate sequences as overhangs next to a gene-specific primer, which are then used for reverse transcription (RT) of mRNA, resulting in first-strand cDNA molecules that are UID-tagged [56]. Alternatively, UIDs can be incorporated in cDNA during the template-switching 5’ RACE protocol [32, 57]. Following UID-tagging, cDNA is used as input for PCR amplification and library preparation can proceed as per usual. After NGS is performed, sequencing reads are grouped together based on their UIDs. Because single- molecule tagging was performed, it is assumed that reads sharing the same UID originated from the same mRNA molecule. Errors can then be corrected by simply building a consensus sequence from UID groups. Another advantage is that counting UIDs instead of raw reads also leads to an improved measurement of transcript abundances (i.e. less bias) [60]. One disadvantage is that incorporation of UIDs increases the size of the amplicon, resulting in additional technological hurdles related to read length. For example, while the Illumina MiSeq platform is able to generate 2x300bp read lengths, the addition of a UID may lead to challenges in obtaining high quality reads across the whole VDJ region because read quality drops towards the end of each read. One way to circumvent this problem relies on adding UIDs to both ends of the amplicon . The subsequent use of Tn5 transposase breaks longer sequences into a mix of sequences that are either anchored at the V-Seqment or the constant region. Non-tagmented amplicons can link both fragments via their UIDs [33]. After sequencing each library variant, the complete read is assembled bioinformatically by building the consensus sequence out of all sequences that carry matching UIDs. However, it has been observed that UID-based consensus building is not able to remove all errors, most likely due mutations that occur early during PCR [32]. Shugay et al. developed a highly effective secondary error-correction step based on read gain/loss filtering, in which clones are completely dropped from the analysis if after consensus building, these clones have lost reads mapping to their original, uncorrected clonal sequence [32].
An additional consideration is that UIDs should be designed to have diversity that is high enough to prevent early saturation, which would lead to tagging different RNA or DNA molecules with the same barcode, a problem often described as the birthday paradox [61,
General introduction
62]. This design can be achieved either by adding sufficiently long, degenerate regions on the reverse primer [32, 34] ,by performing a second-strand cDNA synthesis (which introduces an additional UID [58]), and/or by also setting a similarity threshold for UID groups [62].
However, increasing the sequence space dedicated to the actual UID also proportionally increases the probability that UIDs will accumulate technical errors themselves [63]. Precise experimental control is also needed, such as limiting material inputs to ensure sufficient oversampling of UIDs in the NGS data. Additionally, the observed error across UIDs is even more dependent on the number amplification cycles during library preparation. To counteract this problem, Khan et al. recently developed a protocol known as molecular amplification fingerprinting (MAF), which tags single-molecule IgG cDNA with reverse UIDs and also adds forward UIDs via the forward primer set (specific for variable framework 1 regions) during multiplex-PCR [34]. MAF uses both UIDs to enable error-correction on the UID regions themselves prior to consensus building on the variable regions. Importantly, MAF also uses the ratio of forward to reverse UIDs to generate a normalization constant, which when applied to Ig-Seq data resulted in nearly absolute correction of the bias introduced by multiplex-PCR.
This study also validated error and bias correction using synthetic spike-ins, providing a clear quantification of the degree of error and bias pre- and post-MAF bioinformatics processing.
Synthetic spike-ins are becoming common tools for validation across various NGS methods [64, 65], it is recommended that they also become a standard in immune repertoire sequencing studies.
High-throughput pairing of variable regions
Initial studies in immune repertoire sequencing relied on bulk populations of lymphocytes.
However, this approach is unable to recover the pairing of variable regions because they are expressed as unique transcripts from separate chromosomes. However, possessing additional functional information about variable region pairing is highly desirable because it can be used for a variety of applications such as mAb discovery, vaccine profiling, and analysis of tumor-infiltrating lymphocytes. This kind of information has led to the development of a number of sophisticated methods that are able to achieve high-throughput sequencing of a paired repertoire. These pairing methods utilize the physical linkage of variable regions prior to sequencing or employ a digital linkage that associates sequences via bioinformatic analysis (see Figure 1). It should also be noted that when synthetic libraries are used (e.g. phage display), a physical linkage between chains typically exists in the display platform. In these cases, depending on read length/coverage desires, the CDR3s of both chains may be captured in a single PCR amplicon providing paired variable region data.
General introduction
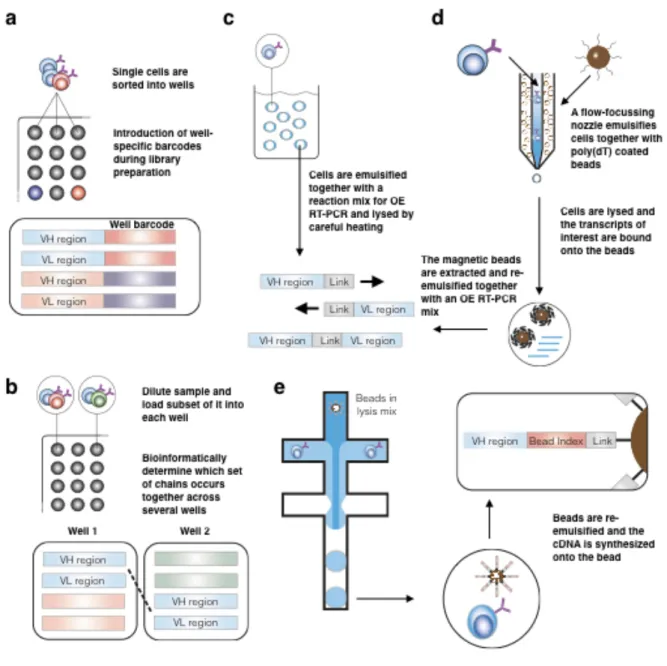
Figure 1-1 Overview of Immune Repertoire Pairing and Sequencing Methods
Overview of immune repertoire pairing and sequencing methods. The common core of many pairing technologies is first to separate the bulk cell population into single-cells and then to pair the sequences of interest (depicted here are variable heavy (VH) and light (VL) regions of the BCR) either via physical or digital linkage. (A) One straightforward approach is to sort single cells into wells on a well plate and introduce well-specific barcodes during library preparation (which is carried out within each of the wells). The resulting sequencing reads can then be linked via this UID [66]. (B) Instead of sorting single cells into each well, aliquots of a larger cell population can be distributed across the plate, and well-specific barcodes can be introduced. Receptor pairing is then performed bioinformatically by determining which set of sequences occurs together across several wells [67]. (C) Another way to separate single cells is to disperse them within water-in-oil emulsion droplets and then lyse the cells within each droplet by heat shock. The resulting sequences are then linked physically by overlap-extension RT-PCR [36].
(D) A more sophisticated approach divides the cell lysis and the RT-PCR step into different compartments. Cells are first emulsified together with poly(dT)-beads and lysis buffer. Transcripts are captured by the beads, which are re-emulsified in a second step. Afterwards the sequences of interest are again linked by RT-PCR [31]. (E) Sequencing by droplet microfluidics (Drop-Seq) utilizes digital linking of not only receptor sequences, but of entire transcriptomes. Here, cells and beads linked to poly(dT) primers (or gene specific primers), which carry a bead- specific UID are encapsulated together in a mix of lysis buffer and RT-reaction buffer. Cells are lysed, and their transcriptome is subsequently reversed transcribed onto the uniquely barcoded bead. Sequencing libraries, which incorporate each barcode are then built directly from each bead [68].
General introduction
Primer matrix pairing
One of the initial steps in generating paired repertoire sequencing data is to devise a strategy that is able to generate libraries from genomic DNA or RNA of single-cells. A straightforward way to achieve this is through flow cytometric sorting of single cells into microwell plates, followed by single-cell library preparation [66, 69, 70] (see Figure 1-1A). In order to increase throughput, each well can be used in conjunction with a specifically barcoded primer set [70], or alternatively, in conjunction with a primer matrix, which utilizes row and column-specific forward and reverse index primers [66, 69]. All of the wells from multiple plates are pooled together, and following sequencing, variable region pairing is identified by their barcode or index sequence. An advantage of this methodology is that sequencing is coupled to the flow cytometry data, thus providing greater resolution of the underlying phenotype of lymphocyte repertoires. As of now, this technology has been able to process up to 4.6 x 104 cells with an efficiency of about 60 % [66, 69]. More recent technologies based on emulsion droplets have pushed the limit of cells that can be processed to even higher numbers (up to 107 cells, see below). However, these gains in throughput have so far come at the expense of collecting flow cytometry data.
Bioinformatic pairing
A simple way to extract pairing information bioinformatically is to link variable regions based on their relative frequency, assuming that naturally paired chains are found in a similar order when ranked by frequency. However, immune repertoires tend to follow power-law distributions, which implies that the majority of cells in a repertoire at a given time are composed of a few dominant clones, whereas the overwhelming majority of clones are present at low frequencies [71]. Because of this phenomena, the above mentioned pairing approach is generally only feasible for a small subset of highly abundant clones [18]. A novel approach, which includes both experimental and bioinformatic elements, has recently been described for high-throughput TCR pairing [67]. In this method small subsets of T cells are separated into microwells, from which sequencing libraries indexed by wells are then bulk prepared, similar to the above mentioned protocols (see Figure 1-1B). After TCR-Seq, a list with all possible Va and Vb combinations within each well is generated. Assuming that the initial cell dilution across all wells was sufficiently high enough, cross-referencing all combinatorial indices elucidates pairings that only occur in combination in the same wells(i.e.
providing Va and Vb pairing). Compared to single cell sequencing, the number of wells from which sequencing libraries have to be prepared is substantially reduced.
General introduction
Emulsion-based pairing
Droplet emulsions are a proven method for capturing and performing reverse transcription (RT) followed by PCR reactions on single-cells. Therefore, the use of emulsions for library preparation in immune repertoire sequencing offers a strategy for high-throughput variable region pairing. A major advantage of emulsions is that they reduce the overall reaction volume and offer straightforward scalability. A simple and cost-efficient approach has been established by Turchaninova et al.; this protocol used water-in-oil droplets to encapsulate T cells, whereby lysis and library preparation were both performed within the droplet [36].
Together with the cell, each droplet also contains a reaction mixture suitable for the RT and PCR steps. The cells are lysed by heat shock, and the variable regions are linked by overlap- extension RT-PCR (see Figure 1-1C). A challenge of this methodology is that the heat shock step may reduce the efficiency of the RT enzyme and the subsequent cDNA synthesis. One way to overcome this problem is to use a methodology that separates the lysis and RT step, as described in a study by Dekosky et al. [30]. In this protocol, B cells are randomly distributed into high-density microtiter chips (60,000 wells/plate), consisting of picoliter wells patterned across a PDMS slide. Additionally, each well is loaded with beads covered with oligo(dT)-primers, which basepair with the 3’ poly(A) tail of (and thus specifically enrich for) mRNA. After the final addition of a lysis buffer the chip is sealed off. Following lysis, the beads are recovered in batch and then encapsulated into single-bead emulsion droplets (see Figure 1-1D). Within each emulsion, cDNA is synthesized and the variable regions are physically linked by an overlap-extension PCR step. This pairing approach was used to identify, with high fidelity, mAbs from the plasmablast compartment of a recently vaccinated human subject [30]. However, the initial encapsulation of cells in the high-density microtiter chip limits the number of cells that can be screened. In a further improvement, a flow-focusing device was used to first encapsulate cells and magnetic poly(dT) beads in a primary emulsion droplet [31]. Cells were then lysed within the droplets and transcripts were again bound to the poly(dT) beads, which were then extracted via magnetic force. Afterwards, the beads were re-emulsified and underwent the same process as in the earlier version of this approach.
Because the updated version does not rely on PDMS slides, the number of cells that can be sequenced increases substantially, up to 106 -107 cells [72].
Combining immune repertoires with single cell transcriptomics
Excitingly, several studies already highlight the expansion of the tools that make up advanced repertoire sequencing. In one example by Han et al., a primer matrix approach for single-cell TCR-Seq was combined with targeted RNA-Seq of T cells [73]. In another example, a newly
General introduction
developed computational tool known as TraCeR was designed to assemble transcripts obtained from single-cell RNA-seq experiments and reconstruct the complete transcriptome of single T cells, also including their TCR variable region sequences [74]. The importance of these methods will only increase as the the price and throughput of commercially available single-cell technologies continues to drop (see Table 1-2). Fan et al. recently established a technology termed CytoSeq, which was able to perform high-throughput gene expression profiling on single-cells [75]. CytoSeq couples microscale plates (patterned with up to 100,000 wells) with magnetic beads which are covalently bound to oligo(dT) primers that carry a particular UID for each bead. Such uniquely barcoded beads are prepared by split- pool synthesis, a concept borrowed from combinatorial chemistry. These microbeads are then separately encapsulated with single-cells in each well while also extracting the transcriptome cells based on the bead UID. So far, CytoSeq has enabled the simultaneous interrogation of transcriptomes from up to 4 x 105 single cells [75]. Two recent studies have established a technology which combines droplet microfluidics with transcriptome sequencing (Drop-Seq) [68, 76] (see Figure 1-1E). Similar to CytoSeq, Drop-Seq uses specifically barcoded microspheres to group sequencing reads coming from the same cell together. To date, these approaches have yet to be specifically applied for immune repertoire studies, but we anticipate that it will only be a matter of time, as the throughput and overall wealth of information provided will lead to substantially greater insight.
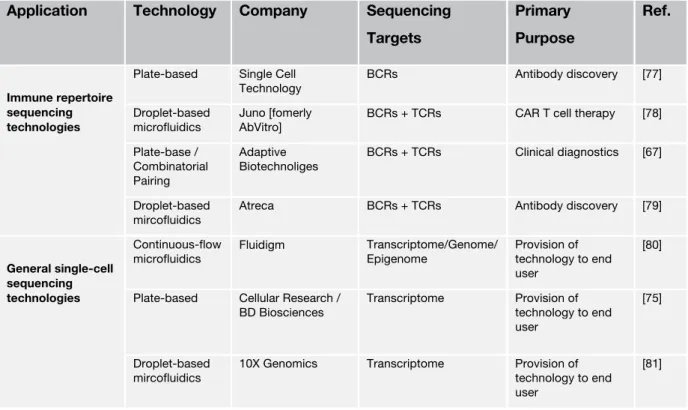
Table 1-2 A brief overview of single-cell sequencing technologies
Application Technology Company Sequencing Targets
Primary Purpose
Ref.
Immune repertoire sequencing technologies
Plate-based Single Cell Technology
BCRs Antibody discovery [77]
Droplet-based
microfluidics Juno [fomerly
AbVitro] BCRs + TCRs CAR T cell therapy [78]
Plate-base / Combinatorial Pairing
Adaptive Biotechnoliges
BCRs + TCRs Clinical diagnostics [67]
Droplet-based
mircofluidics Atreca BCRs + TCRs Antibody discovery [79]
General single-cell sequencing technologies
Continuous-flow
microfluidics Fluidigm Transcriptome/Genome/
Epigenome Provision of technology to end user
[80]
Plate-based Cellular Research / BD Biosciences
Transcriptome Provision of technology to end user
[75]
Droplet-based
mircofluidics 10X Genomics Transcriptome Provision of technology to end user
[81]
General introduction
Concluding remarks and future directions
The advanced methods in immune repertoire sequencing described in this chapter demonstrate the capability to obtain accurate (error and bias-corrected) and variable region paired data. These capabilities have set a new standard for the field of immune repertoire sequencing. While the technologies established for error and bias correction were largely focused on ex vivo biological repertoires, similar approaches may be adapted to synthetic repertoires (e.g. phage, bacteria, and yeast display [82, 83]). Bias is less of a concern in synthetic libraries, since they typically use a single forward and reverse primer. However, the application of error correction (e.g. use of UIDs) is still relevant to synthetic repertoires. Using error corrected data for synthetic libraries enables reliable tracking of clonal enrichment, identification of conserved versus non-conserved amino acids, and decreases the likelihood of overestimating library diversity. Since the faults and limitations of TCR- and Ig-Seq are now well established, researchers can ensure high quality results and interpretations using these methods. However, gaps still remain and in order to gain further insight into lymphocyte function, it may be beneficial to link immune repertoires with transcriptional or epi-genomic data [74] (see Figure 1-2). This high-dimensional information may lead to the ability to predict TCR or antibody specificity and functionality based on transcriptional profiles and refined phenotypic markers.
Figure 1-2 Single-cell sequencing and immune receptors
A simplified example highlighting how single-cell sequencing can link information about a specific immune receptor and different transcriptional profiles. Each single cell exhibits unique levels of transcriptional activity (left- hand side). In reality, one sequencing run can measure the expression level of many thousand genes.
Dimensionality reduction techniques such as principle component analysis or t-SNE can aid visual inspection of these datasets. Here, principle component analysis orthogonally projects the data points onto a lower dimension, which minimizes the (squared) distances between the original points and their projection (right-hand side). This projection reveals cells of similar transcriptional activity and subsets of potentially different cell populations, while the information about each cell’s immune receptor (here pictured in grey) illuminates the developmental lineages of lymphocytes across these compartments.
General introduction
As more advanced methods continue to be established, increased reliability will lead to widespread usage of immune repertoire sequencing for a variety of healthcare applications.
These applications are already being commercialized for characterizing tumor infiltrating lymphocytes, tracking of residual disease in blood-borne cancer, and mAb engineering. The ability to interrogate millions of sequences enables the selection of monoclonal antibodies not just based on binding, but selection of candidates that are more developable (e.g., removing candidates with undesirable T cell epitopes, chemical degradation hotspots).
Overall, it is expected that advanced immune repertoire sequencing will lead to more efficient and fast development of immunological therapeutics and diagnostics.
This chapter is an author produced adaptation of a peer reviewed research article published in Frontiers of Immunology in 2018.
Synthetic standards combined with error and bias correction improve the accuracy and quantitative resolution of antibody repertoire sequencing in human naïve and memory B cells
Simon Friedensohn*, John M. Lindner*, Vanessa Cornacchione, Mariavittoria Iazeolla, Enkelejda Miho, Andreas Zingg, Simon Meng, Elisabetta Traggiai, andSai T. Reddy
*equal contribution
Author contributions:
S.F., J.M.L., E.T., and S.T.R. designed experiments. V.C. performed B-cell enrichment, sorting, and mRNA extraction. M.I. and S.F. prepared IgH libraries. J.M.L. designed primer sequences. J.M.L. and A.Z. designed antibody spike-ins. A.Z., S.M., and M.I. conducted preliminary experiments. S.F. was responsible for the bioinformatics pipeline and analysis.
S.F. and J.M.L. analyzed data and prepared figures. S.F., J.M.L., E.T., and S.T.R. wrote the paper.
Published in:
Front. Immunol., 20 June 2018 | https://doi.org/10.3389/fimmu.2018.01401
Abstract
High-throughput sequencing of immunoglobulin repertoires (Ig-Seq) is a powerful method for quantitatively interrogating B cell receptor sequence diversity. When applied to human repertoires, Ig-Seq provides insight into fundamental immunological questions, and can be implemented in diagnostic and drug discovery projects. However, a major challenge in Ig- Seq is ensuring accuracy, as library preparation protocols and sequencing platforms can introduce substantial errors and bias that compromise immunological interpretation. Here, we have established an approach for performing highly accurate human Ig-Seq by combining synthetic standards with a comprehensive error and bias correction pipeline. First, we designed a set of 85 synthetic antibody heavy chain standards (in vitro transcribed RNA) to assess correction workflow fidelity. Next, we adapted a library preparation protocol that incorporates unique molecular identifiers (UIDs) for error and bias correction which, when applied to the synthetic standards, resulted in highly accurate data. Finally, we performed Ig- Seq on purified human circulating B cell subsets (naïve and memory), combined with a cellular replicate sampling strategy. This strategy enabled robust and reliable estimation of key repertoire features such as clonotype diversity, germline segment and isotype subclass usage, and somatic hypermutation (SHM). We anticipate that our standards and error and bias correction pipeline will become a valuable tool for researchers to validate and improve accuracy in human Ig-Seq studies, thus leading to potentially new insights and applications in human antibody repertoire profiling.
Introduction
Adaptive immune responses are governed by cooperative interactions between B and T lymphocytes upon antigen recognition. A hallmark of these cells is the somatic generation of clonally unique antigen receptors during primary lymphocyte differentiation. In particular, B cell antigen receptors (BCRs, and their analogous secreted form, antibodies) are formed upon rearrangement of the incomplete, germline-encoded variable (V), diversity (D, heavy chain only), and joining (J) gene segments. Unique V(D)J recombination events in differentiating B cells create a highly complex repertoire of receptors (generally interchangeably referred to as BCR, antibody, or immunoglobulin (Ig) repertoires), which is shaped upon antigen experience to produce the more targeted, high-affinity memory BCR network. In-depth and accurate characterization of these repertoires provides valuable insight into the generation and maintenance of immunocompetence, which can be used to monitor changes in immune status, and to identify potentially reactive clones for therapeutic or other uses. Due to rapid technological advances, high-throughput sequencing of Ig genes (Ig-Seq) has become a
major approach to catalog the diversity of antibody repertoires [84-86]. Ig-Seq applied to human B cells has potential in a variety of applications [87], particularly in antibody drug discovery [88-90], profiling for vaccine development [91, 92], and biomarker-based diagnostics [93, 94]. Additionally, Ig-Seq is enabling a more comprehensive understanding of basic human immunobiology, for example by ascertaining B cell clonal distribution and repertoire complexity across physiological compartments in health and disease [95, 96].
A major challenge when performing Ig-Seq is the production of accurate and high-quality datasets. Several current library preparation protocols are based on target enrichment from genomic DNA or mRNA [97]. For example, the conversion of mRNA (more commonly used due to transcript abundance and isotype/subclass identification) into antibody sequencing libraries relies on a number of reagents and amplification steps (e.g. reverse transcriptase, multiplex primer sets, PCR), which potentially introduce errors and bias. Due to the highly polymorphic nature of repertoires especially from affinity-matured memory B cells and plasma cells, it is essential to determine if such technical noise occurs at non-negligible rates, as this could alter quantitation of critical repertoire features such as clonal frequencies, germline gene usage, and somatic hypermutation (SHM) [97, 98]. One way to address this is by implementing synthetic control standards, for which the sequence and abundance is known prior to sequencing, thus providing a means to assess quality and accuracy [99].
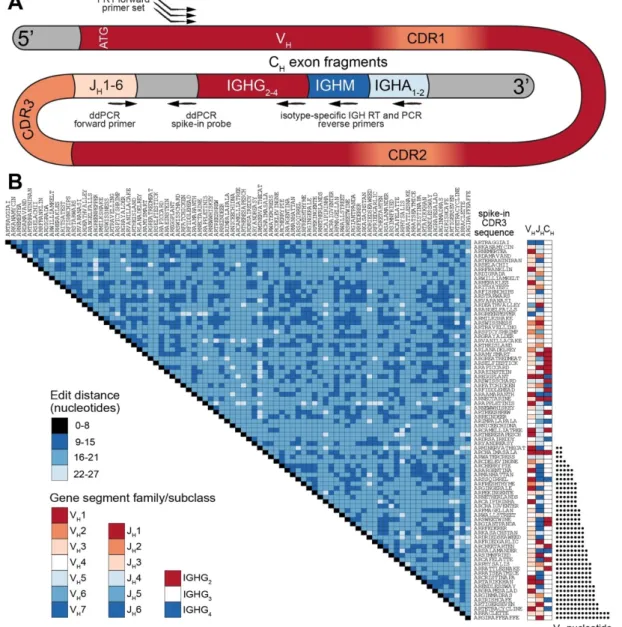
Several examples of Ig-Seq standards have already been presented; Shugay et al.
sequenced libraries prepared from a small polyclonal pool of B and T lymphocyte cell lines, and observed nearly 5% erroneous reads, resulting in approximately 100 false-positive variants per clone [100]. Recently, Khan et al. developed a set of synthetic RNA (in vitro transcribed) spike-in standards based on mouse antibody sequences, which were used to show that a substantial amount of errors and bias are introduced during multiplex-PCR library preparation and sequencing [101].
Various experimental and computational workflows exist to mitigate the effects of errors and bias in Ig-Seq. One of the most advanced and powerful strategies is to incorporate random, unique molecular identifiers (UIDs, also commonly referred to as UMIs or molecular barcodes) during library preparation. Following sequencing, error correction can be performed by clustering reads that share the same UID to form a consensus; reads sharing the same UID are assumed to be derived from the same original mRNA/cDNA molecule [102]. Furthermore, by counting the number of UIDs (instead of total reads), one can correct for cDNA abundance bias [103, 104]. Several iterations of UID-tagging have been developed for Ig-Seq, such as UID labeling during first- and second-strand cDNA synthesis [105], UID addition during RT template switching [106], and so-called “tagmentation” of UID-labelled amplicons [107].