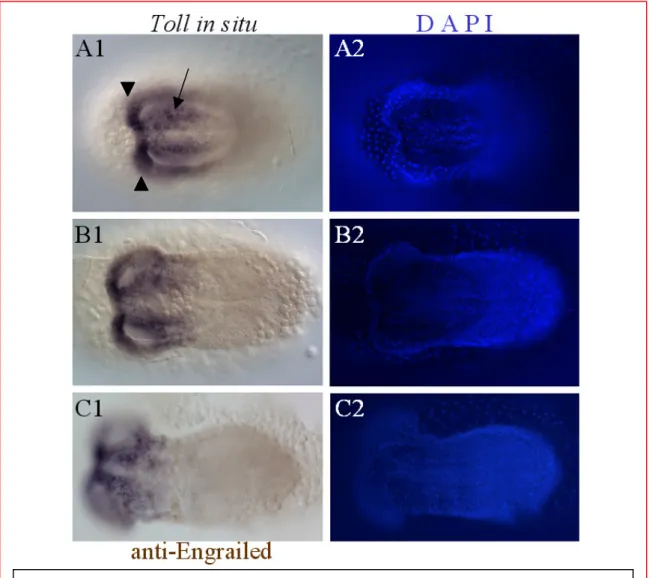
Funktionelle Studien zur dorsoventralen Musterbildung in Tribolium castaneum
108
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
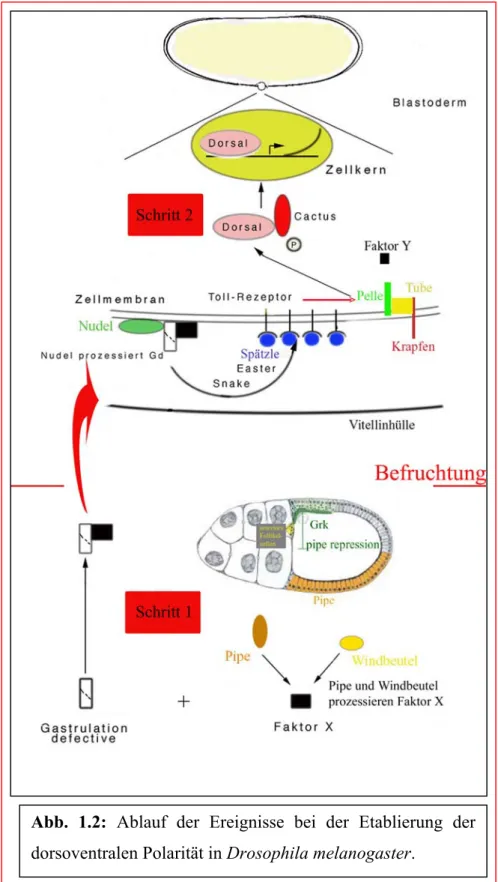
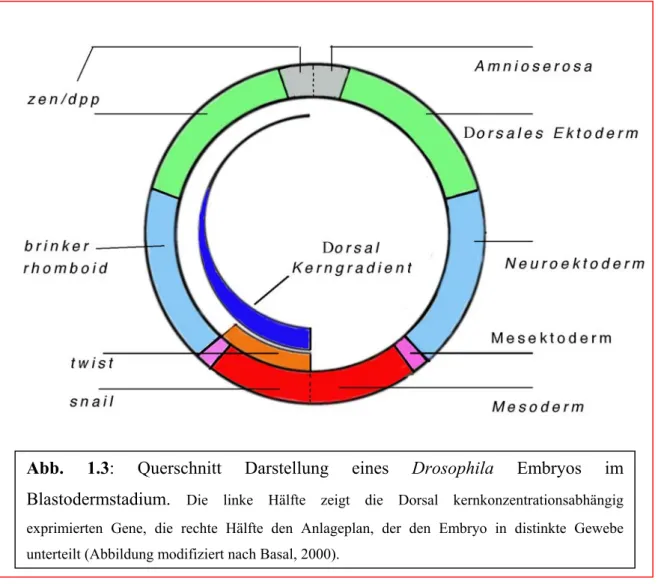
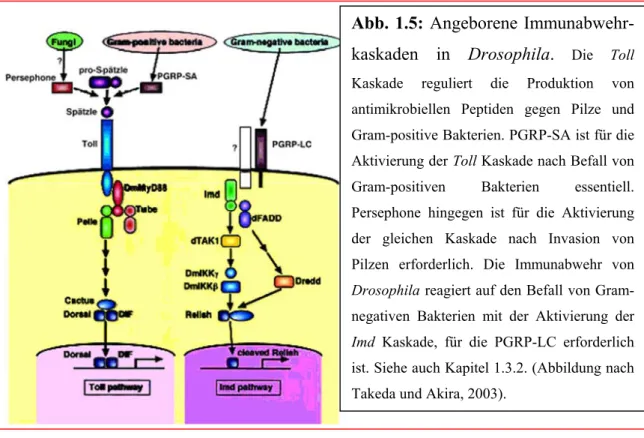
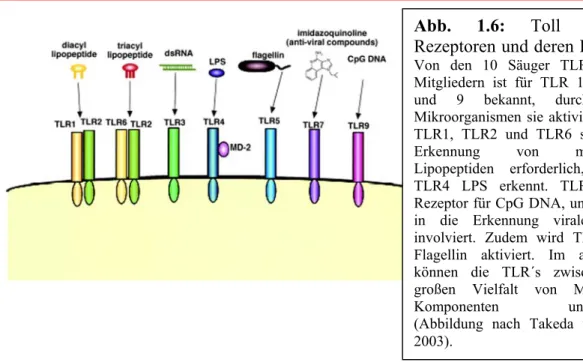
Abbildung
+7

ÄHNLICHE DOKUMENTE
Zwar handelt es sich bei der HEK 293T-Zelllinie nicht um eine lymphatische Linie, sie wurde jedoch trotzdem verwendet, unter der Annahme, dass LKLF in humanen Zellen prinzipiell
Wenngleich eine direkte Phospho- rylierung aufgrund der biochemischen Unzug¨anglichkeit von Prf1 in vivo bisher nicht nach- gewiesen werden konnte, zeigte sich in genetischen
Zu der strukturellen und funktionellen Bedeutung des MARV Glykoproteins wurden bereits ausgedehnte Untersuchungen durchgeführt, wobei die cytoplasmatische Domäne des
Dies spielt bei Erkrankungen mit einer Eosinophilie eine große Rolle, da hier bereits in der Vergangenheit gezeigt werden konnte, dass die Apoptose von Eosinophilen atopischer
These analyses revealed that the genes Tc-rx, Tc-chx, and Tc-six3 play important roles in different steps of the larval brain midline specification and Central
In order to identify the alkene biosynthetic mechanisms, the fatty acid profile was explored in glands and different developmental stages of Tribolium castaneum with
Da die Signale von Fgf8 und Shh auch für die Entwicklung der dopaminergen Neuronen wichtig sind und sie deren Entwicklung sogar induzieren können, ergab sich der Schluss, dass
In order to identify more candidates of the highly conserved core of anterior bilaterian patterning genes, I systematically analyzed the expression and function
