1. Einleitung
1.1 Definition, Historisches, Einbettung in Nachbarwissenschaften
Der Begriff Bio-Anorganische Chemie scheint zunächst widersprüchlich, legt man die historische Definition der Anorganischen Chemie als der Chemie der „Toten Materie“
zugrunde. Allerdings wurde dieses Verständnis der Anorganischen Chemie bereits 1828 durch die Wöhler’sche Harnstoffsynthese ad absurdum geführt. Unter der Bioanorganischen Chemie versteht man heute diejenige Teildisziplin der Biochemie (= Chemie der lebenden Organismen), der sich mit der physiologischen Rolle von anorganischen Elementen und den molekularen Grundlagen ihrer Funktion befasst.
Die Bioanorganische Chemie wird ist etwa 1960 als eigenständiges Teilgebiet akzeptiert. Voraussetzung dafür, dass sich dieses Teilgebiet als eigenständige Disziplin entwickeln konnte, war die Erkenntnis, dass typische anorganische
Elemente in physiologischen Systemen weit verbreitet sind und eine Schlüsselrolle bei zahlreichen physiologischen Funktionen spielen. Beispielsweise enthält etwa die Hälfte aller bis heute bekannten Enzyme mindestens ein Metallion als wesentliche, für dessen Funktion meist sogar unabdingbare (essentielle) Komponente.
Maßgeblich für die Erkenntnis der physiologischen Rolle anorganischer Elemente waren Fortschritte bei den Isolations- und Reinigungsverfahren der Biochemie und die Entwicklung immer empfindlicherer Nachweismethoden, mit denen sich auch Spuren von Elementen nachweisen lassen. Dies ist insbesondere ein Verdienst der Atomabsorptionsspektroskopie (AAS) und der Atomemissionsspektroskopie (AES).
Eine wichtige Triebkraft bei der Entwicklung dieser Disziplin waren auch die
Bemühungen um die Aufklärung biochemischer Reaktionsmechanismen und deren Nutzbarmachung in vitro. Dies schließt die Synthese einfacherer Modellsysteme und die Erforschung ihrer Reaktivität ein. Hier stehen die Biochemie und die
anorganische Chemie in intensiver gegenseitiger Wechselbeziehung. Beispielsweise werden spektroskopische Daten strukturell bekannter Modellsysteme mit denen der physiologisch vorkommenden Systeme verglichen, um detaillierte Informationen zum Aufbau der „anorganischen“ Teilstrukturen im physiologischen System zu erhalten.
Wie der Name schon sagt ist die Bioanorganische Chemie ein hochgradig
interdisziplinäres Gebiet, das mittlerweile fast schon „Modecharakter“ hat. Neben der
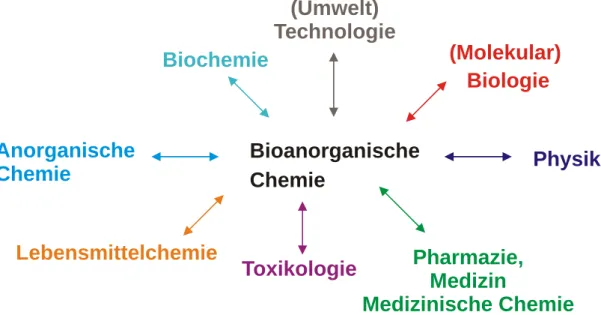
Biochemie und der Anorganischen Chemie gibt es Berührungspunkte zu einer Reihe anderer Teildisziplinen (s. Abb. 1).
Bioanorganische Chemie
Physik (Molekular) Biologie
Pharmazie, Medizin
Medizinische Chemie Toxikologie
Anorganische Chemie
Biochemie
Lebensmittelchemie
(Umwelt) Technologie
Abbildung 1. Die Bioanorganische Chemie und ihre Nachbardisziplinen
Es ergeben sich folgende Berührungspunkte:
- Physik: Bereitstellung und Weiterentwicklung der Nachweis- und
Untersuchungsmethoden
- (Molekular)Biologie:
Bereitstellung physiologischen Materials und dessen gezielte gentechnologische Manipulation
- Toxikologie: Untersuchung des Schadstoffpotentials anorganischer
Verbindungen und dessen molekularer Grundlage
- Pharmazie, Medizin, Medizinische Chemie: medizinischer Einsatz
anorganischer Substanzen in der Diagnostik und der Therapie
- Lebensmittelchemie, Agrar- und Ernährungswissenschaften: Rolleanorganischer Verbindungen für Wachstum und Gedeihen eines Organismus, Schadstoffproblematik
- Technologie, Umwelttechnologie: Bakterieller Stoffabbau in Kläranlagen oder
Sedimenten, Schadstoffabbau und Entgiftung
1.2 Die physiologischen Funktionen von Metallionen
In lebenden Organismen finden sich z. T. größere Mengen an Metallionen. Erste Erkenntnisse über deren physiologische Rolle sammelte bereits Liebig (1803-1873) mit seinen Untersuchungen zum Stoffwechsel anorganischer Nahrungsbestandteile und zum Einsatz von Düngemitteln zur Ertragssteigerung (Nitrate, Phosphate, Kaliumsalze). Als besonders auffallende bioanorganische Verbindungen wurden die Blatt- und Blutfarbstoffe von Seiten der damals noch als Teil der Organischen Chemie begriffenen Naturstoffchemie untersucht (man denke in dem
Zusammenhang an die Blutlaugensalze!). In der Folgezeit lernte man auch, dass einige Krankheiten oder Missbildungen auf mangelnde Versorgung mit bestimmten Spurenelementen zurückzuführen sind. Dabei kann man in für einen Organismus absolut lebensnotwendige, essentielle und dem Gedeihen förderliche (benefitial) Elemente unterscheiden. Die Abgrenzung liegt darin, dass das vollständige Fehlen essentieller Elemente schwere, irreversible Schäden auslöst. Vielfach stellt man fest, dass als förderlich oder essentiell anerkannte Elemente in höherer Konzentration (Dosis) umgekehrt schädigend oder gar toxisch wirken. Ein Paradebeispiel dafür ist Selen. Einmal wurde Selenocystein als 21. essentielle Aminosäure erkannt und werden selenhaltige Präparate in der Nahrungsmittelergänzungsindustrie als
Wunderheilmittel dargestellt, andererseits ist es in größeren Mengen aufgenommen stark toxisch. Darin dokumentiert sich dass Paracelsus’sche Prinzip von der
ambivalenten Wirkung vieler Stoffe. Für ein jedes essentielles oder förderliches Element ist demnach ein Dosis-Wirkungsdiagramm zu formulieren wie Abb. 2 es zeigt.
Konzentration (Dosis) positiv
negativ
Tod Tod
Abbildung 2. Dosis-Wirkungsdiagramm
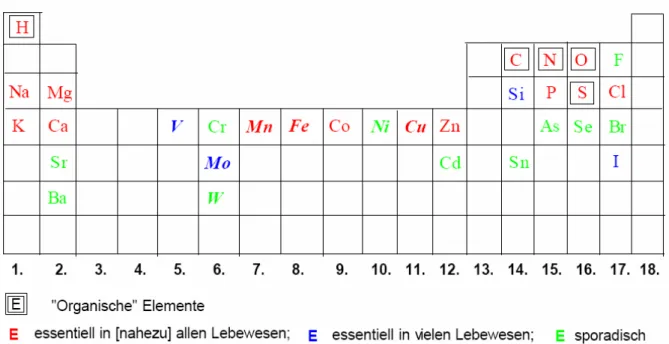
Tabelle 1. Durchschnittlicher Metallgehalt des Menschen
Tabelle 1 gibt eine Übersicht über den durchschnittlichen Metallgehalt im
menschlichen Körper nach Emsley,1 während Abb. 3 die essentiellen Elemente
zusammenfasst. Unter den Übergangsmetallen besitzen fast alle Elemente der 3d-
Reihe eine essentielle physiologische Funktion, während unter den Elementen der
4d-Reihe lediglich Mo eine derartige Rolle spielt.
Funktion Eigenschaft Beispiele Strukturbildung- und
Stabilisierung
Ladung, Bildung schwer löslicher Verbindungen
Ca2+: Skelettaufbau; Ca2+, Mg2+: Stabilisierung der Struktur von DNA; Zn2+:
Stabilisierung der Struktur DNA-erkennender Proteine; Zinkfinger, P,O,C, Si, S, F:
Bestandteile von strukturstabilisierenden Anionen Reizleitung,
Signalübertragung
Ladung, hohe Beweglichkeit Na+, K+: Elektrochemische Potentialsprünge, z.B. bei der Nervenreizleitung, Steuerung der Muskelkontraktion, erfordert Ionenpumpen zur Erzeugung und Ionenkanäle zum Abbau von Konzentrationsgefällen.
Säure-Base Katalyse Lewis-Acidität Mg2+, Zn2+: Katalyse von Hydrolysen oder Kondensationsreaktionen (Phosphatasen, Carboanhydrasen, Hydrolasen etc.), zentrale Rolle beim Auf- und Abbau organischer Verbindungen
Elektronentransport Redoxaktivität Fe/S-Cluster, Fe/Mo-Cofaktoren der Nitrogenasen, Mn-Cluster der Photosynthese, Fe-Porphyrinkomplexe der Cytochrome, blaue Kupferproteine
Sauerstofftransport bei der Atmung
labile Koordination von O2 Fe-haltige Häm-Proteine, Cu-haltige Hämocyanine
Aktivierung kleiner Moleküle, Atomübertragungsreaktionen
Erzeugung und
Stabilisierung radikalischer Zwischenstufen
O-Übertragung: Fen+: Cytochrome; Fen+, Cun+, Con+, Mon+, Vn+, Mnn+: Oxygenasen, Oxidasen, Reduktasen, Ni2+: Hydrogenasen; Übertragung von Alkylresten,
Umlagerungsreaktionen: Co2+: Cobalamine (Vitamin B12) Tabelle 2. Physiologische Funktion anorganischer Bestandteile
Die Funktion der essentiellen Elemente wird deutlich, wenn man sich die
Mangelerscheinungen vor Augen führt, die ihr Fehlen nach sich zieht. Die wichtigsten Befunde sind in Tabelle 3 zusammengestellt.
Tabelle 3. Übersicht über Mangelerscheinungen
Element Mangelerscheinung
Ca Wachstumsbeeinträchtigung (Skelett)
Mg Krampfneigung Fe Anämie, Störung des Immunsystems
Zn Hautschäden, Zwergwuchs, verzögerte sexuelle Reifung Cu Arterienschwäche, Leberstörungen, sekundäre Anämie Mn Unfruchtbarkeit, gestörter Skelettaufbau
Mo Verlangsamtes Zellwachstum, Kariesneigung
Ni Wachstumsverzögerung, Dermatitis
Cr Glukose-Intoleranz (Symptome wie bei Diabetes) Si Störungen des Skelettwachstums
F Karies
I Schilddrüsenfehlfunktion (Kropf), verlangsamter Metabolismus
Se Muskelschwäche, Bluthochdruck
Prinzipiell kann man zwischen Metalloproteinen und Metallenzymen unterscheiden:
In Metalloproteinen treten Metalle als Cofaktoren von Proteinen auf. Metalloenzyme
sind Metalloproteine mit katalytischen Funktionen. Auffällig ist, dass einige der
redoxaktiven Metallionen in biologischen Systemen in eher ungewöhnlichen
Oxidationsstufen vorkommen: Beispiele sind Fe(V), Mn(III), Cu(I), Mo(IV), W(IV),
Co(I), Ni(I), Ni(III).
2. Koordination, Transport und Speicherung von Metallionen
2.1 Bioliganden
Neben einfachen oder komplexen Phosphaten, Sulfid- und Sulfationen, Wasser und dessen deprotonierten Formen (OH
-, O
2-) stehen Metallionen in Enzymen die
folgenden Bioliganden zu Verfügung:
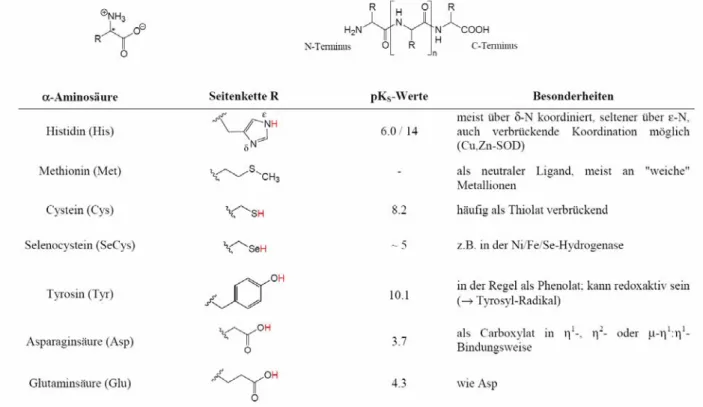
a) Aminosäuren und Peptide (s. Übersicht)
Aminosäuren können Metallionen über ihre Carboxylatfunktion binden. Dabei sind die in Abb. 4 gezeigten, unterschiedlichen Koordinationsmodi möglich. Abb. 5 gibt ein Beispiel für die µ, η
1: η
1-Koordination einer synthetischen Aminosäure.
C
O O
M η1, syn C
O O
M
η1, anti
C
O O
M η2
C
O O
M M
µ,η1:η1 Ca2+ Fen+, Mnn+
'
Abbildung 4. Koordinationsmodi von Aminosäuren an Metallionen
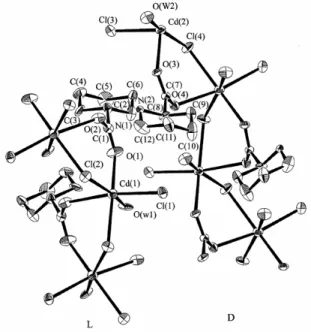
Abbildung 5. Die Struktur von CdCl2(H2O)(DL-Hpip) mit µ, η1: η1-Koordination der Aminosäure
Analog ist eine Koordination am C-Terminus von Peptiden möglich. Bei Polypeptiden
ist jedoch zu berücksichtigen, dass viele Aminosäuren Substituenten mit
Ligandfunktionen in ihrer Seitenkette tragen. Da jedes Peptid nur einen C-Terminus gegenüber einer Vielzahl von derart koordinierenden Aminosäuren aufweist, ist eine Bindung von Metallionen an die Seitengruppen bevorzugt. Tabelle 4 gibt eine
Übersicht über potenziell koordinierende Aminosäuren; ein Beispiel für die Koordination eines synthetischen histidinfunktionalisierten Äquivalents eines Dipeptids gibt Abbildung 6.
Tabelle 4. Aminosäuren/Peptide als Liganden
In Abhängigkeit vom Metallion und seiner Oxidationsstufe existieren gewisse
Präferenzen bezüglich des Aminosäurepartners, der Koordinationszahl und der
Koordinationsgeometrie (Selektivität) (s. Tabelle 5)
Tabelle 5. Charakteristische Bindungspartner und Koordinationsgeometrien für bestimmte Metallionen
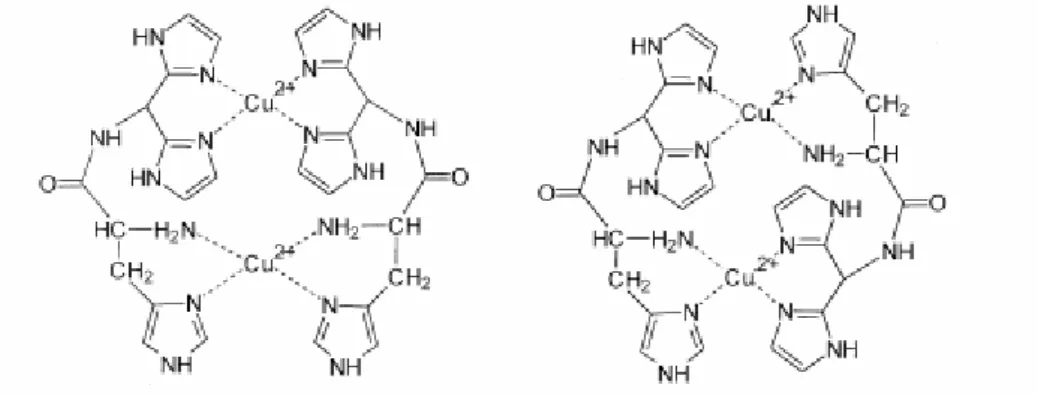
Abbildung 6. Koordination von Aminosäuren mit histidinhaltigen Seitenketten an Cu2+
(Sovago et al, Inorg. Chim. Acta, 339, 2002, 373).
Abbildung 7. Beispiele für Komplexe mit chelatisierender und N-terminaler Koordination von Glycin, s. Mann et al. Dalton Trans. 2007, 1500.
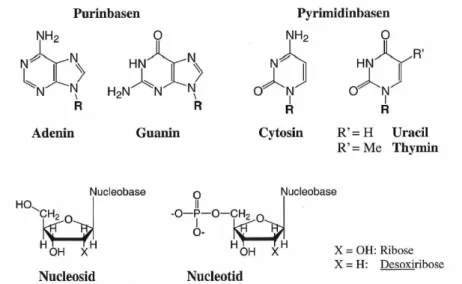
b) Nucleobasen, Nucleotide und Nucleinsäuren (s. Übersicht)
Abbildung 8. Nucleobasen,. Nucleoside und Nucleotide.
Nucleobasen (Abb. 8) sind ambidente Liganden, d. h. sie verfügen über mehrere ungleiche Koordinationsstellen. Je nach der Art (Größe, Ladung) des koordinierten Metallions, der anderen Liganden (H-Brücken-Bindungen) und den äußeren Bedingungen (pH-Wert) können sie als ein- oder mehrzähnige Liganden fungieren und über Imino-, Amino-, Amido-,
Hydroxo- oder Oxofunktionen koordinieren. Beispiele für röntgenographisch gesicherte Strukturen von Metallkomplexen mit Koordination der Nucleobasen Adenin, Cytosin und eines sich von Guanin ableitenden Nucleosids zeigt Abbildung 9.
Pair of asymmetric units of Cu(MIDA)(AdeH)(H2O)] ×/H2O showing the intra -molecular interligand hydrogen bond in both molecules (open dashed lines) and the inter -molecular p,p-stacking interaction between five- and six- membered rings of AdeH ligands (black dashed lines).
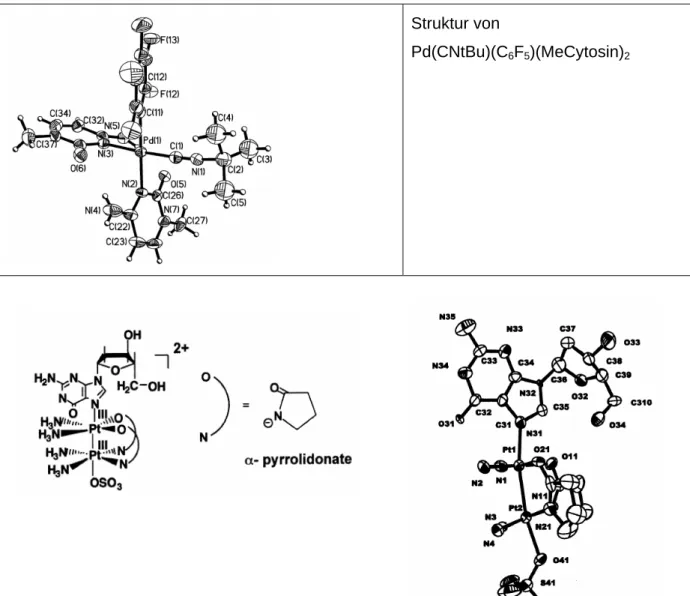
Struktur von
Pd(CNtBu)(C6F5)(MeCytosin)2
Abbildung 9. Verschiedene Beispiele für röntgenographisch charakterisierte Komplexe mit koordinierten Nucleobasen.
Auch ein Beispiel für die chelatisierende Koordination von Pd(II) an einfach deprotoniertes Thymin ist mittlerweile bekannt (Abb. 10).
1Die Synthese erfolgte gemäß:
[(Aryl)
2Pd(µ-OH)
2Pd(Aryl)
2](NBu
4)
2+ 2 1-Methylthymin → 2 [(Aryl)
2Pd(1-
Metyhlthymid)] + 2 H
2O (Aryl = C
6F
3H
2, C
6F
5).
Abbildung 10
Für zytostatisch wirkende Halbsandwich-Ru(II)-Komplexe des Typs (η
6- Aren)Ru(en)Cl] wurde die selektive Bindung an N7 von Guaninbasen in DNA- Oligomeren nachgewiesen. Modellkomplexe zeigen attraktive π-Wechselwirkungen zwischen ausgedehnten π-Arenliganden und dem nichtkoordierten, elektronenarmen Sechsring der Nukleobase sowie stabilisrende Wechselwirkungen über H-Brücken (Abb. 11).
2Ein derartiges „π-stacking“ bewirkt auch den Einschub sog.
„Intercalatoren“ in die DNA, was bei Zytostatika wie cis-Platin-Derivaten und bei der
Fluoreszenzmarkierung von DNA eine wichtige Rolle spielt.
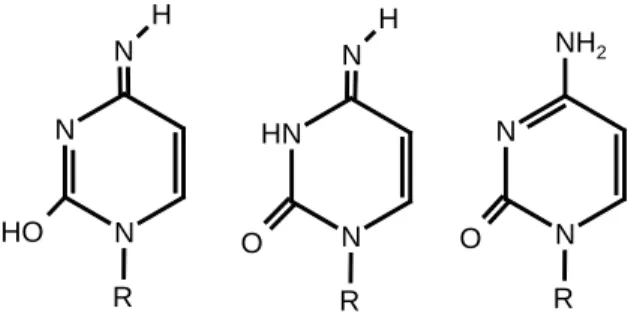
Die Tatsache, dass Nucleobasen in verschiedenen tautomeren Formen vorliegen können, stellt eine zusätzliche Komplikation dar (Abb. 12).
N N
N H
HO R
N HN
N H
O R
N N
NH2
O R
Abbildung 12. Verschiedene Tautomere des N(1)-substituierten Cytosins
Durch Koordination an Nucleobasen können Metallionen mit der Basenpaarung interferieren, indem sie die „falschen“ Tautomeren und „falsche“ Basenpaare stabilisieren. Ausbleibende Reparatur zieht eine Verfälschung der genetischen Information nach sich. Die mutagene und carcinogene Wirkung einiger Metallionen basiert auf diesem Mechanismus. Andererseits kann man diese Koordination auch gezielt medizinisch in der Tumorbekämpfung nutzen (cis-Platin).
c) Makrozyklische N-Donorliganden: Tetrapyrrolliganden und Verwandte (s.
Übersicht, Abb. 13)
NH N
N N
Corrin
Abbildung 13
Die makrozyklischen Chelatliganden können eine breite Vielfalt von Metallionen koordinieren und bilden sehr stabile Komplexe. Dies lässt sich schon daraus ablesen, dass Sedimente, Kohle und Erdöl hohe Konzentrationen an derartigen Komplexen enthalten. Die Makrozyklen haben einen zentralen Hohlraum von genau definierter Größe, etwa 60-70 pm. Komplexe des Mg
2+-Ions (72 pm, Chlorophyll), von Fe
2+/3+in verschiedenen Spin-Zuständen (h. s. Fe
2+: 78 pm, zu groß, l. s. Fe
2+: 65 pm, passt; h. s. Fe
3+: 65 pm, passt, l. s. Fe
3+: 55 pm, zu klein, durchschnittlich 65 pm, Häm-Proteine, Cytochrome), Co
2+(65 pm, Cobalamin) und Ni
2+(69 pm, F-430, Tunichlorin, chlorophyllartiger Ni-haltiger Komplex bei Manteltieren) haben wichtige biologische Funktionen. Gewisse Anpassungen an größere Metallionen sind dadurch möglich, dass diese außerhalb der Ringebene koordinieren oder der Makrozyklus durch Wölbung (doming), Verdrillung (ruffling), Einnehmen einer sattelförmigen (saddling) oder wellenförmigen Struktur (waving) seine Planarität einbüßt. Abb. 14 zeigt diese Verzerrungen zusammen mit den Energien der entsprechenden out-of plane IR-Schwingungen. Diese Verzerrungen werden maßgeblich von der
Aminosäuresequenz zwischen dem Porphyringerüst und einem weiteren axialen Liganden (dem sog. proximalen Liganden) sowie in verknüpften Porphyrinen durch die Aminosäuresequenz der Verknüpfung beeinflusst und sind in verwandten Enzymfamilien weitgehend konserviert.
3Abbildung 14. Deformationsmodi des Porphyrinrings und die Energie der charakteristischen out-of-plane IR-Bande
auch als „Pigmente des Lebens“ bezeichnet. Ihre Redoxaktivität macht diese Komplexe zu essentiellen Komponenten bei den wichtigsten biologischen
Energieumwandlungsprozessen: der Atmung und der Photosynthese. Selbst nach der Koordination an den Makrozyklus verbleiben am Metallion noch zwei weitere Koordinationsstellen. Diese können einmal zur Substratbindung und zum anderen zur Steuerung der Reaktivität durch den trans-ständigen Liganden (trans-Effekt) herangezogen werden. In den Metalloproteinen der Häm-Familie ist eine axiale Position von einem proximalen Histidinliganden besetzt.
d) Ionophore
Dabei handelt es sich um vielzähnige Chelatliganden der Zähnigkeit von ≥ 6, die Ionen mit oft großer Selektivität binden. Ihre Hauptfunktion besteht im Ionentransport.
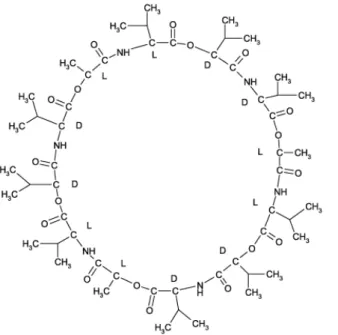
Daher üben sie auch bei der Aufnahme von Ionen aus dem umgebenden Medium und ihrer Speicherung eine wichtige Funktion aus. Ein wichtiger Ionophor für K
+und Ca
2+ist das Valinomycin, ein cyclisches Dodecadepsipeptid, das sich aus Valin und Isobuttersäure her leitet (Abb. 15). Ein weiteres Beispiel ist Nonactin, ein
makrozyklischer Ester, der ebenfalls K
+und Ca
2+bindet (Abb. 16).
Abbildung 15. Valinomycin (links) und Nonactin (rechts)
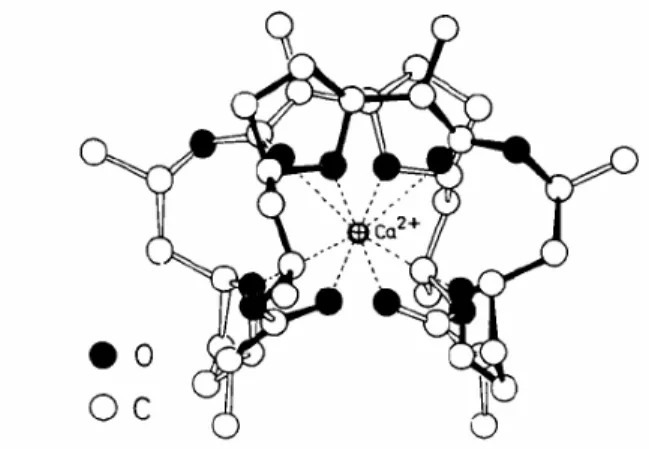
Abbildung 16. Struktur des Ca2+-Komplexes von Nonactin4
In den strukturell charakterisierten K
+und Ca
2+-Komplexen (s. Abb. 16) wird das Metallion von allen Estercarbonyl- und Ether-Sauerstoffatomen koordiniert (KZ = 8).
Infolge der Koordination drehen sich alle acht Donorfunktionen ins Komplexinnere.
An der Außenseite sind die unpolaren Alkylgruppen angeordnet. Dies erlaubt beispielsweise den Transport polarer, schwach koordinierender, hydrophiler und meist substitutionslabiler Metallionen durch die lipophile Doppelschicht biologischer Membranen. Weiter bewirkt die Anhäufung chiraler Zentren eine spezifische
Rezeptorerkennung. Dafür ist auch maßgeblich, dass von den zahlreichen möglichen Konformationen des freien Liganden oft nur eine einzige für die Metallkomplexierung optimal ist. Diese natürlichen Ionophore sind Teil des chemischen Abwehrsystems von Pilzen, Flechten und Meeresorganismen. Ihr gezielter Einsatz stört den
Ionenhaushalt und die Membranfunktion von Bakterien, beeinträchtigt aber die komplexeren Ionentransportmechanismen höherer Organismen kaum. Derartige Makrozyklen werden daher auch medizinisch als Antibiotika eingesetzt.
Eine weitere wichtige Gruppe von biogener Ionophore sind die Siderophore (=
Eisenträger). Dabei handelt es sich um Chelatliganden mit einem Molekulargewicht bis ca. 1500 Da und hoher Spezifität für Fe(III). Wichtige Beispiele zeigt Abb. 17. Ihre Funktionen sind die Mobilisierung, der Transport und die Speicherung von Eisen.
Organismen müssen sich derartiger Liganden bemühen, weil die Konzentration von
Fe
3+im Meerwasser (3⋅10
-7Gewichtsprozent, Fe(OH)
3↓), wesentlich geringer ist alsdie physiologische Konzentration (6⋅10
-3Gewichtsprozent). Auch an Land ist Fe
3+Abbildung 17. Wichtige biogene Siderophore.
Mikroorganismen (Bakterien, niedere Pilze) und viele Pflanzen (insbesondere Wasserpflanzen) vermögen mittels der Siderophore, die sie in das umgebende wässrige Medium abgeben, aus schwerlöslichen Eisenhydroxid-Depots Fe
3+durch Komplexierung zu mobilisieren. Die Fe-bindenden funktionellen Gruppen sind Catecholate (bei den Enterobaktinen), Hydroxamate (bei den Ferrioxaminen und Ferrichromen), Carboxylate und Hydroxycarboxylate (z.B. Rhizoferrin). Die Komplexe sind mehr oder weniger globulär gebaut und verfügen über eine Peripherie aus hydrophilen Gruppen (Amid- und Esterfunktionen), die Wasserlöslichkeit und den Transport im aquatischen System gewährleisten.
Bis heute sind etwa 200 biogene Siderophore von Pilzen, Bakterien und Hefen bekannt. Ihre Biosynthese wird über ein DNA bindendes und von Fe(II) aktiviertes Regulationsprotein (Fe uptake regulation, fur) durch das Angebot an Eisen reguliert.
Ein prominenter Vertreter ist das Enterobactin, ein Tris(catecholat)-Ligand (Abb. 14).
Die Komplexbildungskonstante K
f= [Fe(Ent)] / [Fe]
3+[Ent
3-] (Gl. 2) beträgt ca. 10
49. Synthetische, den Siderophoren nachgebildete Chelatliganden werden auch
medizinisch eingesetzt. Dies beruht zum einen auf ihrer antibiotischen Wirkung.
Mikroorganismen bedürfen für ihre Vermehrung einer kontinuierlichen Versorgung
mit Eisen. Sie sind allerdings nicht dazu in der Lage, dieses aus fest gebundenem
werden, beugt man durch Gabe von mit dem Urin ausscheidbaren Chelatliganden einer Eisenvergiftung vor.
2.2 Transport und Speicherung von Ionen am Beispiel des Eisens
Speicherung und Freisetzung
Um Fe
3+aus der Umgebung aufnehmen zu können, synthetisieren Lebewesen Siderophore. Pflanzen nutzen dazu oft symbiotisch lebende Mikroorganismen. Der jetzt lösliche Komplex wird spezifisch in die Zelle aufgenommen, wobei er mit
Membran-Rezeptoren wechselwirkt und über einen energieverbrauchenden Prozess ins Innere der Zellen transportiert wird. Das Herauslösen des Eisens aus dem
Komplex kann auf mehreren Wegen erfolgen: Hydrolyse des Liganden, Reduktion von Fe
3+zu Fe
2+, das weitaus labilere Komplexe mit diesen Liganden bildet, oder Verdrängung des zur Aufnahme und zum Transport verwendeten Liganden durch einen spezifischen intrazellulären Eisen bindenden Rezeptor. Eine Hürde bei dem Reduktionsmechanismus sind die außerordentlich niedrigen Potenziale der
Fe
3+/Fe
2+-Paare in den Siderophorkomplexen. Dies ist eine direkte Konsequenz der hohen Komplexstabilitäten, wie die folgenden Überlegungen zeigen:
Gemäß der Nernst-Gleichung ergibt sich das Redoxpotential des Gleichgewichtssystems [Fe(Sid)] + 3H
++ e
-⇄ Fe
2++ H
3Sid (3) zu E = E
0+ 0.059V ⋅ log [FeSid] [H
+]
3/ ([Fe]
2+·[H
3Sid]) (4).
Mit K
f= [Fe(Sid)] / ([Fe
3+]·[Sid
3-] (2) und K
s= [H
+]
3[Sid
3-] / [H
3Sid] lässt sich Gl. 4 zu E = E
0+ 0.059V · log [Fe
3+]/{[Fe
2+]}·K
s·K
f} (Gl. 5) umformen.
Dementsprechend ist zu erwarten, dass das Redoxpotenzial des Fe
2+/3+- Paares E
0mit zunehmender Komplexbildungskonstante sinkt, ähnliche pK
S-Werte der Liganden
vorausgesetzt. Dies ist auch der Fall (Tabelle 6)
Tabelle 6. Stabilitätskonstanten und Redoxpotenziale für Fe-Komplexe verschiedener Siderophore
Siderophor log K
f(Fe(III)) E
0in V (pH = 7) Ligand-Typ
Mugineinsäure 18.1 - 0.102 Carboxylat, Amino-N
Aerobactin 22.5 -0.336 Hydroxamat, Carboxylat
Coprogen 30.2 -0.447 Hydroxamat Desferrioxamin B 30.5 -0.468 Hydroxamat
Ferrichrom A 32.0 -0.448 Hydroxamat
Enterobactin ca. 49 -0.790 Catecholat
Alterobactin A 51±2 -0.972 Catecholat, Hydroxy,
Carboxylat
Für Alterobactin A (Alt A) zeigen potentiometrische Titrationen, dass bei Fe
3+-
Bindung sechs Protonen freigesetzt werden. Mit Hilfe der NMR-Spektroskopie konnte gezeigt werden, dass je zwei Protonen von der Catecholateinheit und den beiden Hydroxyaspartat-Untereinheiten abgegeben werden. Der Fe
3+-Alterobactionkomplex ist über einen pH-Bereich von 4 bis 9 hydrolysestabil. Komplexbildung stabilisiert Alt A gegen Hydrolyse. In Abwesenheit des Metallions wird Alt A bei pH = 8 mit einer Halbwertszeit von 11.6 h unter Spaltung des Lactonrings zum offenkettigen Alt B hydrolysiert. Der Komplex ist unter den gleichen Bedingungen dagegen stabil.
5Demnach ist ein gekoppelter Protonierungs-/Reduktionsmechanismus
wahrscheinlich. Ist Fe
2+erst einmal in der Zelle freigesetzt worden, muss es wegen
seiner Labilität direkt verarbeitet oder gespeichert werden.
Abbildung 18. Alterobactin A und Alterobactin B und Voltammogramm des Fe(III)-Komplexes
Die Speicherfunktion übernehmen hochspezialisierte Proteine, insbesondere das Ferritin und das Hämosiderin. Im Ferritin bilden 24 gleichartige Protein-
Untereinheiten eine Hohlkugel mit einem Außendurchmesser von etwa 130 Å und einem Innendurchmesser von 75 Å, in die ca. 1200 Fe-Atome (max. 4500 Fe)
eingelagert werden (Abb. 19). Die ungefähre Zusammensetzung des Fe-Kerns lautet Fe(III)
9O
9(OH)
8(H
2PO
4) mit wechselndem Phosphat-Anteil. Aus EXAFS-Daten kann man schließen, dass die Fe-Atome von sechs O-Atomen im Abstand von etwa 195 pm und von etwa 7 Fe-Atomen im Abstand von ca. 330 pm umgeben sind. Er ähnelt dem metastabilen Mineral Ferrihydrit 5 Fe
2O
3·9 H
2O (= Fe
10O
6(OH)
18. Dieses weist eine hexagonal dichteste Packung von O-Atomen auf. Die Oktaeder sind über
gemeinsame Kanten zu Doppelsträngen verknüpft. Die Doppelstränge sind ihrerseits durch gemeinsame Ecken mit Nachbarsträngen verbunden (Abb. 20).
6Die
Einlagerung und Freisetzung des Eisens erfolgt als Fe(II).
Abbildung 19. Schematischer Aufbau des Ferritins. Die Cn-Achsen markieren Kanäle n- zähliger Symmetrie. Jede Untereinheit besteht aus vier langen α-Helices, die zu einem Bündel gruppiert sind. Sie werden von einer 5. kurzen α-Helix (symbolisiert durch E) verbunden. N symbolisiert den gegenüberliegenden N-Terminus.
Abbildung 20. Die vermutliche Struktur von Ferrihydrit.
Die Fe
3+-Ionen sind durch µ-O und µ-OH-Brücken miteinander verknüpft. Zur Proteinwandung hin erfolgt die Ankopplung über Carboxylatgruppen (von Glutamat und Aspartat). Das Eisen befindet sich im high-spin Zustand; das gegenüber
isolierten high-spin Fe
3+- Ionen (spin-only Wert S = 5/2, 5.92 µ
B) deutlich
verminderte magnetische Moment von nur 3.85 µ
Bweist auf antiferromagnetische Kopplung hin. Für den An- und Abtransport des Eisens dienen Kanäle im
Proteinmantel, die einen Durchmesser von ca. 10 Å haben. Beim Einbau wird das
Eisen zunächst an der nach außen weisenden Seite des Kanals als Fe
2+koordiniert.
Nach Transport ins Innere und Ankopplung an die Carboxylatgruppen der inneren Wandung oder den dort bereits gebundenen Fe
2O
3-Keim erfolgt oxidative Addition von O
2. Dabei gebildete zweikernige, peroxoverbrückte (Fe
3+)
2-Intermediate bilden durch nachfolgende weitere Oxidation und Hydrolyse zwei FeO(OH)-Zentren. Die Nettogleichung für diesen Prozess lautet 4 Fe
2++ O
2+ 6 H
2O → 4 FeO(OH) + 8 H
+. Bemerkenswerterweise erfolgt der Einbau von Fe
3+-Ionen mit einer Geschwindigkeit von bis zu 3000 Fe-Atomen pro Sekunde.
Für den Transport von Fe-Ionen nach außen werden zwei mögliche Mechanismen diskutiert. Zum einen könnten unpolare Reduktionsmittel durch die hydrophoben Ferritin-Kanäle vierzähliger Symmetrie nach innen gelangen und Fe
3+reduzieren. Die Fe
2+-Ionen würden anschließend durch hydrophile Kanäle dreizähliger Symmetrie nach außen transportiert. Alternativ könnten Reduktionsmittel an der Außenseite des Ferritins andocken und Elektronen durch die Proteinhülle ins Innere abgeben.
Eventuell wird die Fe(III) Reduktion in beiden Fällen durch Fe(II)-Chelatliganden unterstützt.
Hämosiderin hat einen noch höheren Speichergehalt als Ferritin, ist aber unlöslich.
Man vermutet, dass es in Lysosomen aus Ferritin entsteht, dessen Proteinhülle teilweise abgebaut wird. Lysosome sind kugelförmige Vesikel, die Proteine, Polysaccharide, Lipide und Nucleinsäuren abbauen können.
Transport
Zum Fe-Transport dienen ca. 80 kDa schwere Proteine der Transferrin-Familie. Ihre
Hauptfunktion besteht darin, Fe-Ionen vom Ort der Resorption, der Speicherung oder
des Abbaus der roten Blutkörperchen zu den blutbildenden Zellen im Knochenmark
zu transportieren. Die einzelne Peptidkette besitzt zwei halbmondförmige Bereiche
mit je einer Bindungsstellen für Eisen. Es wird dort von zwei Tyrosinat-, einem
einzähnigen Aspartat, einem Histidin und einem chelatisierenden Carbonatliganden
gebunden. Die Bindung des Carbonats erfolgt simultan zu der des Eisens. Der
Proteins. Abb. 21 zeigt die offene Struktur von Pferdelactoferrin in der Apoform und die geschlossene Struktur der beladenen Form.
Abbildung 21. Die röntgenographisch bestimmte Struktur von Pferdelactoferrin
Abbildung 22. Die Koordinationsumgebung um die Fe-Ionen im Transferrin; links:
schematisch, rechts: reale Struktur der Fe-Bindungseinheit in der N-Domäne des humanen Transferrins
Die Stabilitätskonstante des Ferritin Fe
3+- Komplexes liegt bei etwa 10
20istg aber stark vom pH-Wert abhängig; bei pH = 5,5 ist K nur noch ca. 10
12und bei pH 4,5 liegt K
stabunterhalb des Wertes für den Citratkomplex. Zur Weitergabe des Eisens an die Zelle wird, so die plausibelste Hypothese, Transferrin an einen Rezeptor angelagert, zum Endosom eingeschnürt und ins Zellinnere transportiert. Hier wird ein pH von 5.0 - 5.5 hergestellt. Das nach Protonierung des Transferrins und Reduktion als Fe
2+freigesetzte Eisen wird dann schnell an den niedermolekularen Eisenpool im Zytosol übergeben und das Apotransferrin wieder in das Plasma abgegeben. Tatsächlich transportiert die Gesamtmenge des körpereigenen Transferrins pro Tag etwa 40 mg Eisen, während die gesamte Aufnahmekapazität bei lediglich 7 mg Fe liegt.
Transferrin-Rezeptoren (TfR) finden sich auf allen sich teilenden Zellen. Die TfR Expression ist koordiniert mit der Zellproliferation, und wird routinemäßig als Maß z.B. für das Wachstumspotential von in-vivo Tumoren herangezogen.
Neben Fe
3+kann Transferrin auch andere dreiwertige Ionen wie Cr
3+und Al
3+, daneben auch Cu
2+, Mn
2+, Co
3+sowie Ti
4+aufnehmen. Tatsächlich bindet Ti
4+mit höherer Affinität an Transferrin als Fe
3+! Der Transport von Ti
4+spielt bei der Mobilisierung dieses normalerweise extrem schwerlöslichen Ions - Ti
4+bildet in wässriger Lösung bereits bei niedrigem pH oxo/hydroxoverbrückte, unlösliche Polymere – eine wichtige Rolle. Letzteres wird für die Korrosion medizinischer Implantate im Körper sowie für die Anreicherung von Ti in malignem Gewebe verantwortlich gemacht. Tumorzellen haben einen erhöhten Eisenbedarf und exprimieren signifikant mehr Transferrin als gesunde Zellen. Der selektive Ti
4+-
Transport in Tumorgewebe ist für Ti(IV)-Zytostatika wie Cp
2TiCl
2von Bedeutung. Der Transport von Al
3+-Ionen durch Transferrin zum zentralen Nervensystem und die dortige Anreicherung dieses Ions, das ja nicht mehr durch Reduktion mobilisiert werden kann, stellt einen wichtigen Aspekt bei dessen Toxizität dar. Al
3+-
Ablagerungen wurden als (Mit)-Auslöser von Krankheiten wie Alzheimer-Krankheit diskutiert, doch ist diese These sehr umstritten.
Das folgende Schema zeigt den Fe-Kreislauf in höheren Lebewesen in vereinfachter
Form.
Unreife Erythrozyten
Hämosiderin [Fe3+] [Fe2+] Häm und Globin Ferritin
Protoporphyrin
Erythrozyten
Hämoglobin [Fe2+] (120 - 150 mg)
(2500 - 3000 mg)
Milz und andere Gewebe Zerstörung der Erythrozyten
[Fe2+] Ferritin [Fe3+]
extrazelluläre Flüssigkeit Fe(III)-Transferrin
[Fe2+] Häm-Enzyme Ferritin [Fe3+]
Hämosiderin [Fe3+]
Myoglobin [Fe2+] Hämoglobin [Fe2+]
Erythrozyten Plasma
Fe(III)-Transferrin
Schweiß, Urin Schleimhaut
[Fe2+]
Ferritin [Fe3+]
Faeces Gastrointestinaltrakt
Nahrung
30 mg/Tag
5 mg/Tag 30 mg/Tag
Leber und andere Gewebe (150 - 200 mg)
Entsprechende Aufnahme-, Speicher- und Transportmechanismen existieren
vermutlich auch für andere Metallionen, doch sind die Prozesse dort bei weitem nicht so genau verstanden wie beim Eisen. Ein Beispiel ist das Vanadium. Seescheiden (Ascidiae), Pilze und Braunalgen haben die Fähigkeit, dieses Element bis zu einem Faktor von 10
7gegenüber ihrer Umgebung anzureichern. Auch dazu bedienen sich diese Organismen bestimmter Ionophore. Aus dem Fliegenpilz wurde beispielsweise der Naturstoff Amavadin isoliert, ein Komplex mit von zwei 2,2’-Oxy(imino)-
dipropionat-Liganden koordiniertem Vanadium der Oxidationsstufe V oder IV
(Abbildung 23). Die Stabilitätskonstante beträgt etwa 10
23.
Abbildung 23. Die Struktur von Amavadin
Vanadium ist Bestandteil der gängiger Nitrogenasen (Reduktion von N
2zu NH
3) und von Haloperoxidasen. Letztere katalysieren die Halogenierung organischer Stoffe unter Mithilfe von H
2O
2und sind Teil des Abwehrsystems dieser Organismen.
In Seescheiden (Ascidien), welche symbiontisch mit bestimmten Cyanobakterien leben, findet sich eine Fülle makrozyklischer Peptide aus modifizierten Aminosäuren.
Beispiele zeigt Abb. 24. Diese können Ionen wie Ca
2+oder Cu
2+selektiv mit mäßig hohen Komplexbildungskonstanten binden. Die Struktur eines zweikernigen,
carbonatverbrückten Kupferkomplexes zeigt Abbildung 24. Auch für diese wird eine Funktion als Ionophore bei der Aufnahme und dem Transport von Cu
2+in marinem Milieu diskutiert. Die Makrozyklen wirken antiviral und antineoplastisch. Ferner konnte nachgewisen werden, dass Cu
2+oder Ca
2+- Ionen die einzelnen
Aminosäurebausteine über einen Templateffekt präorganisieren und die selektive
Makrolaktamisierung begünstigen. Insofern wird auch diskutiert, dass die Metallionen
eher für die Biosynthese der Makrozyklen wichtiger sind als die Makrozyklen für den
Ionentransport.
7Literatur
1. Ruiz, J.; Villa, M. D.; Rodíguez, V.; Cutillas, N.; Vicente, C.; López, G.;
Bautista, D., Inorg. Chem. 2007, 46, 5448.
2. Chen, H.; Parkinson, J. A.; Parsons, S.; Coxall, R. A.; Gould, R. O.; Sadler, P.
J., J. Am. Chem. Soc. 2002, 124, 3064.
3. Jentzen, W.; Ma, J.-G.; Shelnutt, J. A., Biophys. J. 1998, 74, 753.
4. Vishwanath, C. K.; Shamala, N.; Easwaran, K. R. K.; Vijayan, M., Acta Cryst.
1983, C39, 1640.
5. Holt, P. D.; Reid, R. R.; L. Lewis, B.; W. Luther, G.; Butler, A., Inorg. Chem.
2005, 44, 7671.