Untersuchungen zum Reaktionsverhalten von Pentelidenkomplexen
249
0
0
Volltext
(2)
(3)
(4)(5)
(6)(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
(42)
(43)
(44)
(45)
Abbildung
![Abbildung 2: Graphische Darstellung der Grenzorbitale von [Cp*P{W(CO) 5 } 2 ].](https://thumb-eu.123doks.com/thumbv2/1library_info/4132209.1552120/19.918.151.812.343.587/abbildung-graphische-darstellung-grenzorbitale-cp-p-w-co.webp)
![Abbildung 4: Ausgewählte Grenzorbitale von Verbindung [iPrNHP{W(CO) 5 } 2 ] (2a).](https://thumb-eu.123doks.com/thumbv2/1library_info/4132209.1552120/32.918.257.633.355.769/abbildung-ausgewählte-grenzorbitale-verbindung-iprnhp-w-co-a.webp)
+7
ÄHNLICHE DOKUMENTE
Die phosphor- und borgebundenen Wasserstoffatome wurden anhand der Restelektronendichte lokalisiert und isotrop verfeinert. Die Koordinaten der übrigen Wasserstoffatome
• der Anspruch, bei der Modellierung ökonomischer Handlungssituationen die Ganzheit- lichkeit kaumännischer Arbeitzusammenhänge zu wahren. Wohlverstanden, sollte diese Liste,
Um diese Relation zu erhalten wurde das totale Differentiale dM (T, H ) benutzt, um dS(T, H) als Differential dS(T, M ) zu schreiben (Alternativ diese Relation auch mit den auf Blatt
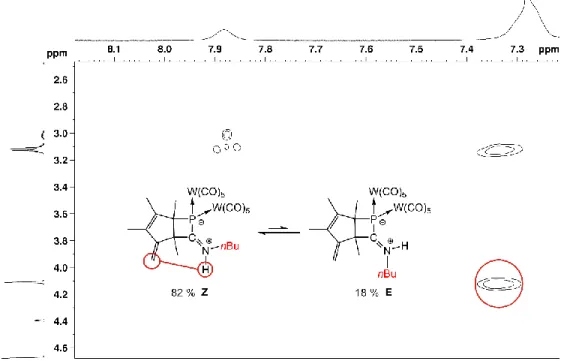
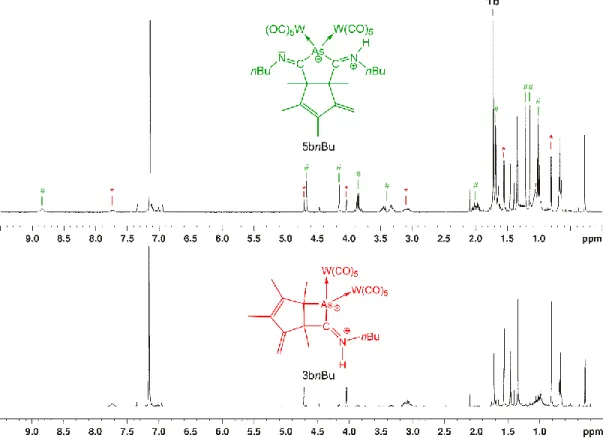
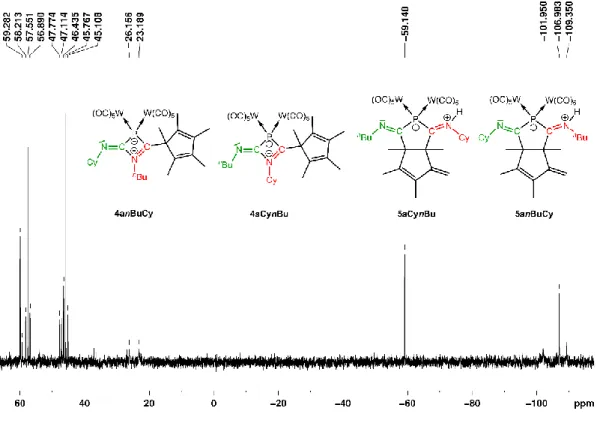
Die anionischen Teilspektren lieferten lediglich zur Matrix gehörende Signale, so daß sich damit keine Aussage treffen läßt, ob es sich bei den Substanzen 9 und 10 um
daß jeder Sludent sich selbst eon 80Id Ober doe Fraktionen mac:I1en VNd und dahf1f ~r haKen es nocht ft:lr "'n\'OIl, hl8f eine gegenS8lbge
In das eine wird Baeyer-Reagenz im Überschuss dazu gegeben, in das andere Bromwasser; beide Reagenzgläser werden gut geschüttelt.. Das Reagenzglas mit dem Bromwasser wird
Die radikalische Substitution sollte vor dem Versuch bereits von den SuS verstanden sein, damit der Versuch auch richtig gedeutet
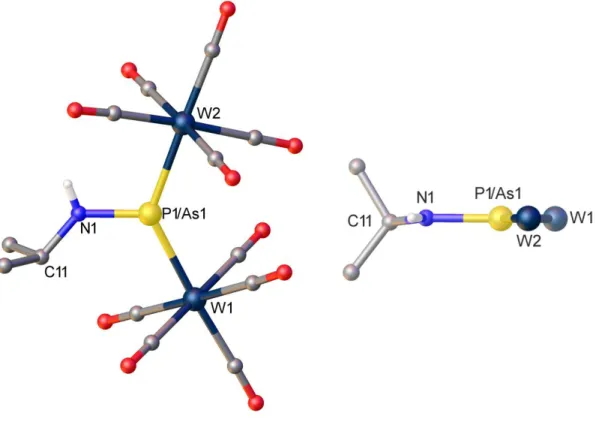
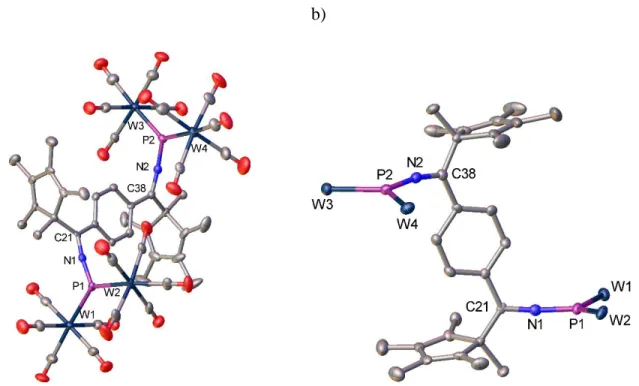
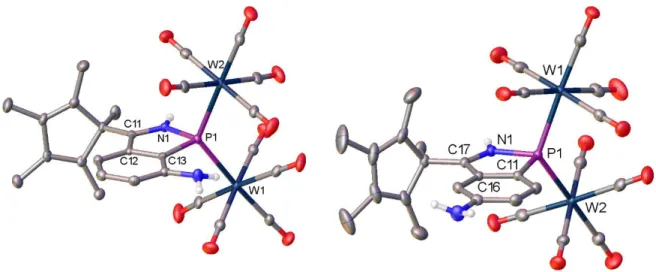
Für eine Röntgenstrukturanalyse geeignete Kristalle von 6 konnten aus einer Lösung in Toluol, solche von 7 aus einer 1:1 Mischung aus Toluol und Hexan bei –28°C erhalten werden..
