W . M . W h i t e G e o c h e m i s t r y Chapter 1: Introduction
19
0
0
Volltext
(2)
(3)
(4)
(6)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
Abbildung

+7

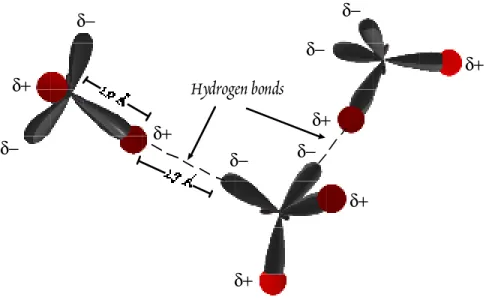
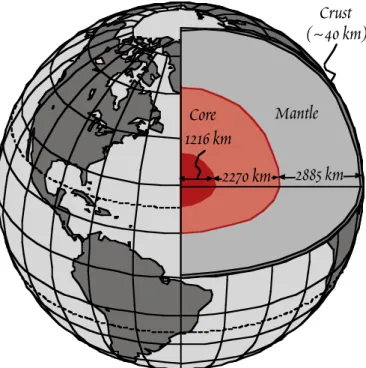
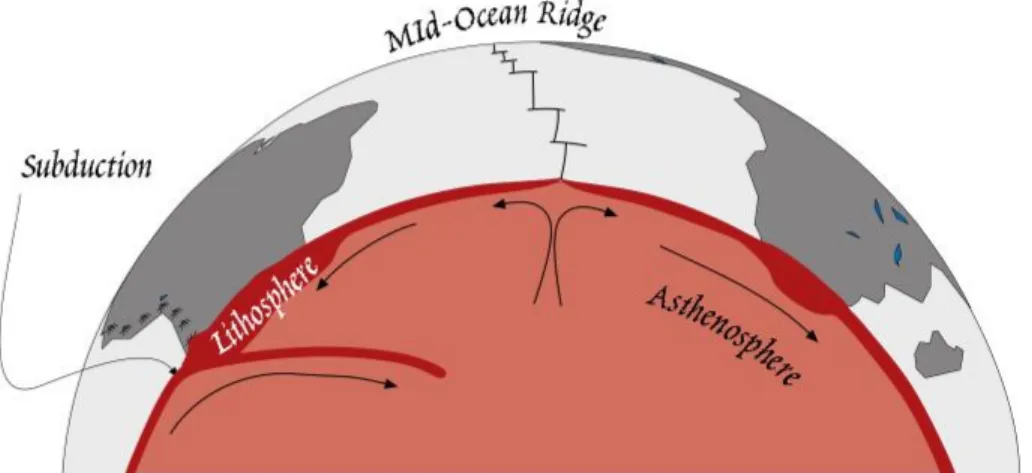
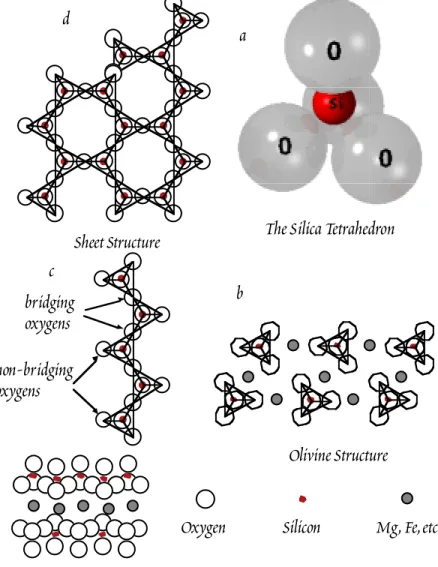
ÄHNLICHE DOKUMENTE
Niemand wird dies mehr ändern können und wenn sie in die Gedanken einer vermeidlichen Drecksau nicht einsteigen wollen, na und – niemand zwingt sie dazu aber es
In view of the successful results of the Federal Office of Energy’s project, Small-scale circulation pumps with a high level of efficiency and the subsequent intensive
On the basis of the research results obtained in the field of electric motors and drives, a Swiss Federal Office of Energy workshop was held at Techno- park at the end of October
I will try to push through the claim that technography is not just one mode among others of literary writing; that all literary writing is in fact technographic, in the sense that
Marie, Soldat, Ignaz, Wagner, Peter Schl., Professor, Major, Wirt, Händler Ein Soldat betritt den Hof und setzt sich an einen Tisch.. --- Marie Grüß‘ Euch Gott,
Papier macht die Schere stumpf und du wirst es beim Zuschneiden deines nächsten Stoffes merken , Der Stoff wird.. dadurch mehr gerissen als
Während ihre Großmutter sich langsam in einer embryonalen Lage zusammenkrümm- te, spürte Solveig, dass es an der Zeit war, gehen zu lernen.. Wenn sie die Arme über ihren Kopf hob
[r]
