Alcohols in the Presence of Metal Salts
Dissertation
Zur Erlangung des Doktorgrades Dr. rer. nat.
an der Fakultät für - Chemie und Pharmazie - der Universität Regensburg
vorgelegt von Andreas Okun aus Perlesreut
Diese Arbeit wurde angeleitet von: Prof. Dr. Oliver Reiser Promotionsgesuch eingereicht am: 09.11.2015
Promotionskolloquium am: 09.12.2015
Prüfungsausschuss: Vorsitz: PD Dr. Rainer Müller
1. Gutachter: Prof. Dr. Oliver Reiser 2. Gutachter: PD Dr. Sabine Amslinger 3. Gutachter: Prof. Dr. Manfred Scheer
Regensburg angefertigt.
Besonders bedanken möchte ich mich bei Herrn Prof. Dr. Oliver Reiser für die Aufnahme in seinen Arbeitskreis, die Überlassung des interessanten Themas, die anregenden Diskussionen und die stete Unterstützung.
Meiner Familie
A Introduction ... 1
1. Organocatalysis – The Beginning of a Great Success Story ... 1
2. Asymmetric Enamine Catalysis ... 3
2.1 Asymmetric Aldol Reactions ... 3
2.2 Asymmetric Mannich Reactions ... 5
2.3 Asymmetric Michael Reactions ... 6
3. Iminium Catalysis ... 8
3.1 Cycloadditions ... 8
3.2 Conjugated Additions ... 9
4. Brønsted Acid Catalysis ... 11
5. Brønsted Base Catalysis ... 13
6. Lewis Acid Catalysis ... 14
B Organocatalysis of L-Proline in the Presence of Metal Salts .... 16
1. Introduction ... 16
1.1 The Nonlinear Effect (NLE) - From the Discovery to Present Research ... 16
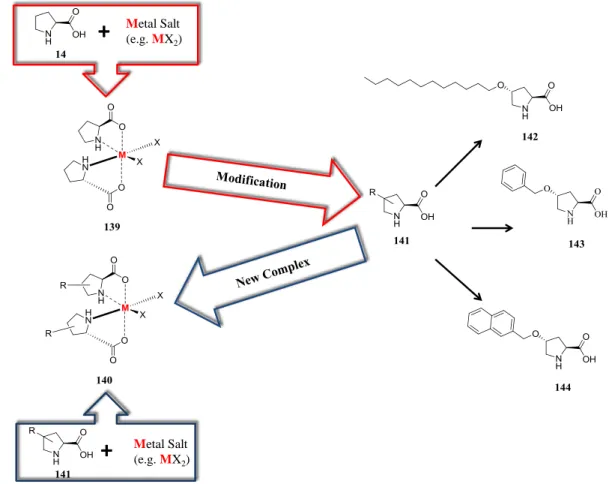
1.2 4-Substituted L-Proline Derivatives... 31
1.3 Synthesis and Applications of the 4-Substituted L-Proline Derivatives 142-144 ... 35
2. Synthesis of Catalysts 142-144 ... 39
3. L-Proline/Metal – complexes as Catalysts in Asymmetric Aldol Reactions ... 42
3.1 Introduction ... 42
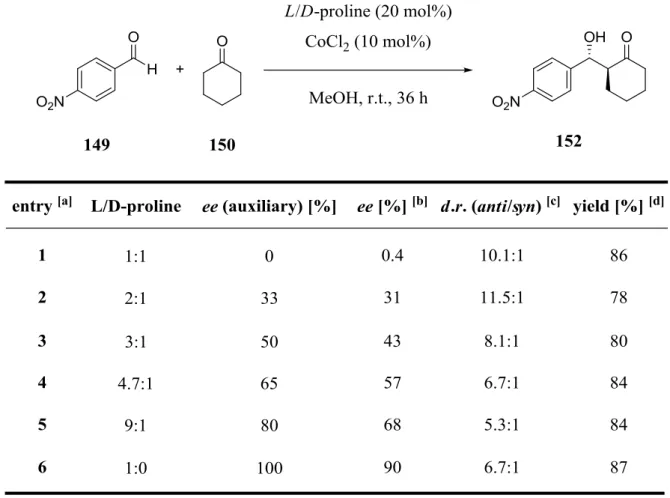
3.2 Investigation of the NLE (nonlinear effect) ... 45
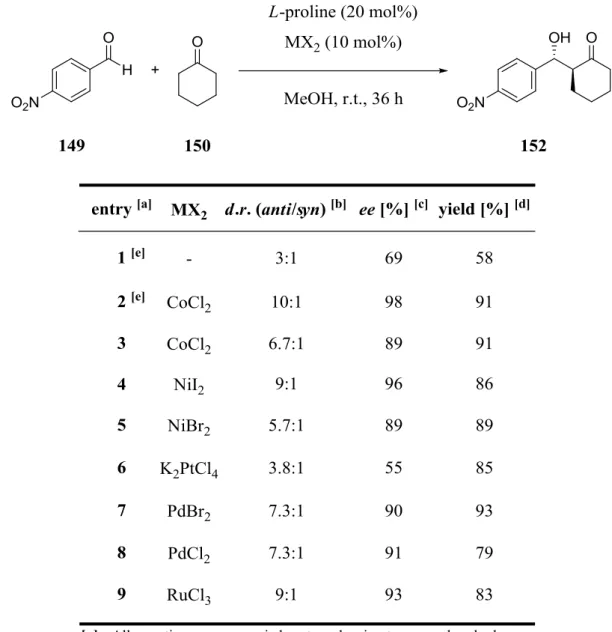
3.3 Screening of Various Metals in the L-Proline Catalyzed Aldol Reaction ... 48
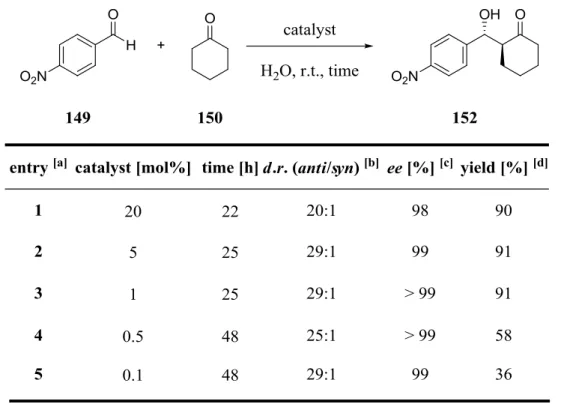
3.4 Optimization of the L-Proline/CoCl2 (2:1) Catalyzed Aldol Reaction ... 51
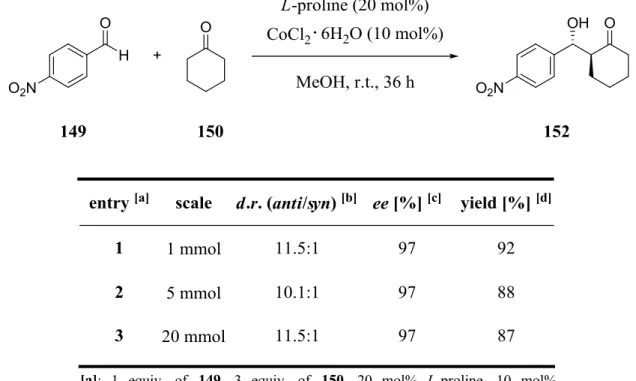
3.5 Upscaling with simplified equipment ... 53
3.6 Evaluation of 4-Substitued L-Proline Derivatives in the Aldol Reaction ... 55
3.7 Summary ... 64
4. L-Proline/Metal-Complexes as Catalysts in Michael Additions ... 65
4.1 Introduction ... 65
5. L-Proline/Metal -Complexes as Catalysts in the Baylis-Hillman
Reaction ... 74
5.1 Introduction ... 74
5.2 L-Proline/Metal-Complexes as Novel Catalysts in the Baylis-Hillman Reaction ... 77
5.3 Summary ... 86
C Organocatalysis of Azabox Ligands in the Presence of CoCl2∙6H2O ... 87
1. Introduction ... 87
1.1 The Discovery of Box Ligands as Versatile Applicable Structure in Catalysis ... 87
1.2 Azabox/CoCl2∙6H2O as Powerful Catalyst ... 89
2. Azabox/CoCl2∙6H2O as Catalyst in the Baylis-Hillman Reaction ... 92
3. Summary and Outlook ... 96
D 1,2-Diamino Alcohols – Synthesis and Applications in Catalysis ... 97
1. Introduction ... 97
1.1 Primary Amines – Often Underestimated Catalysts with Unique Properties ... 97
1.2 Acyclic 1,2-Amino Alcohols as Versatile Structural Motive in Catalysis ... 100
1.3 1,2-Amino Alcohols – Easy Accessible Structures ... 101
2. Novel Synthesis Strategy for 1,2-Diamino Alcohols ... 107
2.1 Library of 9 novel 1,2-Diamino Alcohols ... 107
2.2 Fine-Tuning Approaches on 1,2-Diamino Alcohol 311 ... 110
2.3 Summary and Outlook ... 112
3. 1,2-Diamino Alcohols as Versatile Structure in Organic Synthesis- Application as Catalysts or Ligands ... 113
3.1 Introduction ... 113
3.2 1,2-Diamino Alcohols as Catalysts in the Michael Addition... 115
3.4 1,2-Diamino Alcohols as Catalysts in the Asymmetric Friedel-Crafts Alkylation... 132
3.5 1,2-Diamino Alcohols as Ligands in the Henry Reaction ... 135
3.6 1,2-Diamino Alcohols as Ligands in the Enantioselective Addition of Diethylzinc to Aldehydes ... 142
3.7 1,2-Diamino Alcohols as Ligands in the Ruthenium-Catalyzed Enantioselective Transfer Hydrogenation of Ketones ... 150
E Summary ... 160
F Experimental Part ... 161
1. Instruments and General Techniques ... 161
2. Synthesis of Compounds ... 163
3. Spectra ... 202
G References ... 236
H Appendix ... 250
1. Curriculum Vitae ... 250
I Acknowledgment – Danksagung ... 252
J Declaration ... 254
A Introduction
1. Organocatalysis – The Beginning of a Great Success Story
Asymmetric synthesis is a method for the preparation of chemical compounds which aims to bias the synthesis in favor of producing one stereoisomer over another stereoisomer.
Therefore, asymmetric synthesis is a wide field in organic chemistry which can be achieved via different methods. The most applied techniques are the use of catalysts from the chiral pool, reactions mediated by chiral auxiliaries, (kinetic) racemic resolutions or asymmetric catalysis (Figure 1).[1] The latter is the most important area in sustainable chemistry.
Therefore, the development of new and more efficient methods with the ulterior motive of waste avoidance, higher atom economy, energy saving and generating high stereoselectivity is of great interest in the field of organic chemistry. The modern asymmetric catalysis is based on three big pillars, namely enzyme catalysis, organometallic catalysis and organocatalysis (Figure 1).[2]
Figure 1. The most important methodes in asymmetric synthesis.
Enzyme catalysis is highly selective but often requires special reaction conditions (e.g. water as solvent) and is limited to certain substrates. Metal catalysis is highly selective but, at the same time, very air or moisture sensitive and the products often contain traces of metals, which in pharmaceutical production is undesirable. Among them, organocatalysis is one of the todays most chosen methods for generating high stereoselectivities in C-C-bond
Moreover, it is suitable to many organic transformations including aldol reactions, Diels-Alder reactions, epoxidations, cyclopropanations, alkylations, oxidations and reductions.
Scheme 1. Catalytic cycles for the main activation pathways in organocatalysis.[2-3]
Generally, there are four different types of organocatalysts, Lewis bases and acids, and Brønsted bases and acids. Organocatalysis is ruled by Lewis base catalysis such as amines (e.g. L-proline (14)) and carbenes. Brønsted acid catalysis has become the second big field in organocatalysis and takes place over a direct hydrogen transfer or H-bond activation. The counterparts, Lewis base and Brønsted base catalysis, are only seldom used in organocatalysis (Scheme 1).[2-3] In the following, the most important activation modes and catalysts will be briefly introduced.
Lewis base catalysis
Lewis acid catalysis
Brønsted acid catalysis
Brønsted acid catalysis
2. Asymmetric Enamine Catalysis
The asymmetric enamine catalysis has become the most important field of organocatalysis in the last years, due to its high atom and step economy. The reactivity is based on the enamine formation which lowers the LUMO (lowest unoccupied molecule orbital) energy, leading to an increased C-H acidity. The enamine catalysis could proceed over two pathways, either a nucleophilic substitution by reaction of 4 with a single bond containing electrophile 5 (e.g. alkyl halides) or via a nucleophilic addition by reaction of 4 with a double bond containing electrophile 9 (e.g. aldehydes, imines, Michael acceptors) (Scheme 2).[4]
Scheme 2. Enamine catalysis by the example of a nucleophilic substitution (left) and addition (right).[4]
In the following, the most important catalytic structures based on this activation mode are presented.
2.1 Asymmetric Aldol Reactions
Aldolases, nature’s catalyst for direct asymmetric aldolizations of unmodified carbonyl compounds are using primary amino groups as Lewis base (e.g. class I aldolases). Inspired by this model, many organocatalysts with an amino acid motif were developed for the aldol reaction, mainly based on L-proline (14). Moreover, also primary amino acids, dipeptides or L-proline-based catalysts were utilized (Scheme 3).[4]Therefore, aldol reactions are one of the best investigated C-C bond forming reactions.
Scheme 3. Examples of organocatalysts for various aldol reactions.
In the 1970s, Hajos-Parrish-Eder-Sauer-Wiechert discovered the first aminocatalytic asymmetric aldol reaction, namely an intramoleculare L-proline (14) catalyzed 6-enolendo aldolization of di- and triketones.[5] Further studies revealed that also primary amines (e.g. (1R, 2S)-cispentacin (15) or (S)-phenylalanine (16)) were prone to catalyze these intramolecular cyclizations even with higher enantioselectivities than those obtained with L-proline (14).[6] The first amine-catalyzed asymmetric direct intermolecular aldol reaction was found by List et al., namely an L-proline (14) catalyzed, asymmetric direct aldol reaction of acetone (130) with different aromatic aldehydes.[7] However, in this case primary amines turned out to be inferior catalysts compared to L-proline (14).[4] Even in the presence of water, an aldol reaction of cyclic ketones to electron-deficient aldehydes was practicable catalyzed by primary amines (e.g. (S)-valine (17) and (S)-alanine (18)) reported by Cordova and co workers in 2005.[8] The first investigations in the field of L-proline-derived amino alcohol amides (e.g. 19 and 20) were done by Gong et al. and showed increased stereoselectivities, but slightly lower yields compared to L-proline (14).[9] The L-proline-derived N-acylsulfonamide catalysts 21 generate superior results compared to L-proline (14) as the sole catalyst and were developed Berkessel and co-workers.[10] Moreover, Zhao et al. reported an aldol reaction in which they used the C2-symmetric ligand 22.[11] Outstanding entantioselectivities were obtained by the application of L-proline-based amides 23 and 24 in
direct aldol reactions of acetone (130) and various aldehydes by Singh and co-workers.[12]
Furthermore, even in the presence of water a direct aldol reaction of ketones and aldehydes was feasible by the use of trans-4-silyloxy-proline (25), discoverd by Hayashi and co-workers.[13] Moreover, Tsogoeva et al. showed the good applicability of dipeptides, containing primary -amino acids (e.g. 26), as catalyst for the asymmetric direct aldol reaction.[14] These manifold examples show the great potential of mainly secondary, but also primary amines to catalyze various aldol reactions with excellent outcome.
2.2 Asymmetric Mannich Reactions
The Mannich reaction is a beneficial three component reaction between two carbonyl compounds and an amine (ammonia, primary or secondary) which give rise to
-amino-carbonyl compounds that are core structures in a great number of drugs and natural products, thus making it to an indispensable tool for organic chemists.[4] The first organocatalyzed Mannich reaction between different ketones, p-anisidine and aldehydes catalyzed by L-proline (14) was discovered by List et al. in 2000.[7a, 15] From that moment on, the interest of many researches was raised to develop new organocatalysts for highly stereoselective three component Mannich reactions, mostly based on L-proline structures (Scheme 4). In 2004, Hayashi et al. showed, that 4-siloxyproline (30) was applicable as catalyst in the asymmetric Mannich reaction to a broader scope of substrates compared to L-proline (14), however with identical results.[16] The DMTC catalyst (31) was the first time used by Barbas and co-workers in the reaction of acetone (130) with different preformed aldimines.[17] Due to its better solubility in different organic solvents, the pyrrolidine-based tetrazole catalyst 32 was applied to this reaction as well.[18] Jørgensen et al. studied the preparation of asymmetric quaternary carbons derived from the reaction of ketimines and unmodified aldehydes using catalyst 33.[19] In 2002, the methoxypyrrolidine catalyst 34 was applied by Barbas and co-workers in the transformation of unmodified aldehydes and N-PMP-protected -amino ethyl glyoxylates.[20] The proline-derived pyrrolidinesulfonamide 35 was developed for the reaction of ketones with -imino esters by Wang et al..[21] Ley and co-workers applied the sulfonamide catalysts 36 and 37 to various Mannich reactions.[22]
Scheme 4. Examples of organocatalysts for Mannich reactions.
Furthermore, not only secondary amines were utilized in the Mannich reaction also primary amino acids like (S)-alanine (18) and (S)-tryptophan (39) and alanine-based tetrazole (38) were used as catalysts.[23] In summary, these examples demonstrate the high applicability of secondary but also primary amine catalysts to diverse asymmetric Mannich reactions.
2.3 Asymmetric Michael Reactions
The asymmetric Michael addition has become an indispensable tool in organic chemistry for generating stereogenic centers. Thus, great effort was spent in developing new and especially atom economic strategies for such transformations. For this purpose, possible methodes are enamine-catalyzed enantioselective additions. The pioneering work in this field was done by Stork et al. which paved the way for a manifold potpourri of different approaches in this direction.[4, 24] In Scheme 5, a few examples of organocatalysts which were applied to the asymmetric Michael addition are summarized.
Scheme 5. Examples of organocatalysts used in asymmetric Michael reactions.
The first examples of L-proline (14) catalyzed asymmetric Michael additions were reported by Yamada and co-workers in 1969.[25] Since then many other examples, intramolecular[26] or intermolecular[15a, 22, 27]
, were published in which L-proline (14) was utilized as catalyst.
Moreover, L-prolinol (43)[28], L-proline-derived diamines 44[29] and pyrrolidine sulfonamide 35[30] were as well applied to this reaction. Furthermore, bifunctional catalysts like the L-proline-derived thiourea catalyst 45 were used in the asymmetric Michael addition of cyclohexanone to nitroolefins.[31] On the other hand, also primary amines found their application in asymmetric Michael additions of ketones and aldehydes to nitroolefins, especially dipeptides like 46[32] or amino amides like 47[33]. Another class of primary amine catalysts are bifunctional primary amine-derived chiral thiourea structures like 48[34], 49[35]
and 50[35b], revealing a great potential compared to secondary amine, proline-based chiral thioureas because of their outstanding activity and reactivity. In summary, so far a great number of primary and secondary amines were used as organocatalysts in the asymmetric Michael addition to generate the products in high yields and stereoselectivities.
All in all, the three presented reaction types are the most important applications in asymmetric enamine catalysis. Beside them, there are also the asymmetric - and -functionalization of carbonyl compounds, which are not covered here (for further information see reference [4]).
3. Iminium Catalysis
In 1864 Schiff[36] discovered the condensation reaction of aldehydes and ketones 12 with primary amines 51 in which the starting materials and the products are in equilibrium (Scheme 6).[37] The position of the equilibrium is strongly depending on the pKa value because the primary amine-derived imines 53 are basic thus they are present as iminium ions 52 in acidic solution.[38]
Scheme 6. Formation of iminium ions 52 and imines 53, if the starting material is a primary amine.
However, by the reaction of secondary amines 51 with aldehydes and ketones 12 only iminium ions 52 can be formed. These latter discoveries were the basis for extensive studies of small organic molecules which were able to catalyze organic reactions especially stereoselective ones via iminium activation. As a consequence of the iminium salt formation 52, the electrophilicity compared to the corresponding aldehyde or ketone 12 is increased, which makes the compound more suitable against a nucleophilic attack.[39] This concept, in modern terms, was described by MacMillan and co-workers in 2000 and was named
“LUMO-lowering catalysis”.[40] This was the motivation for many working groups to devote great effort in the development of new synthesis strategies based on iminium-catalyzed processes. In the following, a brief summary of the most important iminium catalyzed transformations and the used organocatalysts is given.
3.1 Cycloadditions
The first organocatalytic cycloaddition was reported by MacMillan and co-workers in 2000.
They applied the imidazolidinone catalyst 54 to the Diels-Alder reaction between activated enals and dienes via iminium ion formation.[40] Moreover, they used this catalyst for a dipolar cycloaddition between Crotonaldehyde and different nitrones.[41]
Figure 2. Examples of organocatalysts used for cycloadditions.
Karlsson and Högberg utilized catalyst 55 to the [3+2]-addition of cyclic aldehydes and different nitrones with much better results than obtained with imidazolidinone 54.[42] Catalyst 57 was used in a [3+2]-addition between enals and cyclic azomethine imines by Chen and co-workers in 2006.[43] The primary amines 57[44] and 58[45] were both utilized in the Diels-Alder reaction with an excellent outcome.
3.2 Conjugated Additions
Conjugated additions are important reactions in organic chemistry, which give access to a great diversity of carbonyl compounds depending on the used nucleophile. Over time, many C-, H-, S-, N- and O-nucleophiles were tested for such addition reactions.[39] In the following a small selection of different organocatalysts used for this approach are shown (Figure 3).The first L-proline (14) catalyzed conjugated addition of nitroalkanes to cycloalkenones was reported by Hannessian and Pham in 2000.[46] Before this discovery Yamaguchi et al. utilized the L-proline salts 59[47] and 60[47d, 48] to the asymmetric Michael addition of malonates or nitroalkanes to ,-unsaturated aldehydes. The chiral imidazolidinone 61 was applied to a Friedl-Crafts alkylation by MacMillan et al.[49] and moreover, was used as catalyst in the key step of a total synthesis of various alkaloids by Banwell and co-workers[50]. The pyrrolidin-based tetrazole 32 was used in the asymmetric addition of nitroalkanes to enones by Ley et al..[51] Moreover, the pyrrolidine-derived catalyst 62 was widely used in diverse asymmetric conjugated additions of malonates[52], thiols[53] or hydroxylamines[54] to
,-unsaturated carbonyl compounds.
Figure 3. Examples of organocatalysts used for conjugated additions.
However, besides secondary also primary amines were applied. The quinine based catalysts 63 and 64 were utilized to a broad scope of asymmetric conjugated additions, most often in the additions of different nucleophiles, like 1,3-dicarbonylcompounds[55], dicyanoalkanes[56]
and indols[57] to ,-unsaturated ketones. Tsogoeva et al. studied the histidine-based dipeptide 26 in the addition of nitroalkanes to cyclohexanone.[58]
There are many other activation modes that proceeding via iminium catalysis. All of them leading to C-C-double bonds or C-N-double bonds either in the product or in the transitions state (for more detailed information see reference [39]).
In conclusion, iminium catalysis has grown to an often used and well established part of organocatalysis. However, the high catalyst loadings required (up to 20-30 mol%) are a certain drawback, thus there is still room for improvement in terms of developing faster and more selective catalysts.
4. Brønsted Acid Catalysis
The field of Brønsted acid catalysis is currently becoming an important pillar of organocatalysis. This type of activation could be divided into two categories. On the one hand hydrogen-bonding catalysts like thiourea 66 and TADDOL derivatives 67 and on the other hand stronger Brønsted acid catalysts like BINOL derivatives 68 and phosphoric acids 69 (Figure 4).[59]
Figure 4. Chiral Brønsted acids.
The binaphthol derivatives 68 were utilized to large variety of reactions including the enantioselective Morita-Baylis-Hillman [60] as well as the enantioselective aza-Morita-Baylis-Hillman[61], the Mannich[62] and the Diels-Alder reaction[63]. Moreover, different BINOL-derived phosphoric acids 69 were applied to miscellaneous reactions like Mannich type reactions[64], aza-Friedel-Crafts alkylations[65], Strecker[66] and aza-Diels-Alder reactions[67] and transfer hydrogenations with Hantzsch ester as hydride source[68].
On the other hand, there are also examples of Brønsted acids catalyzing reactions via hydrogen bonding (Figure 5). Even though, L-proline (14) is usually considered to belong to the group of enamine catalysts it is also possible to assign it to the group of hydrogen bonding catalysts because of its carboxylic acid moiety which could additionally activate the substrate via H-bonding. This concept was first presumed by Barbas et al. in the L-proline (14) catalyzed asymmetric aldol reaction[7b] and later Wu and co-workers achieved improved results by installing a dual H-bond donor instead of the carboxylic acid in catalyst 19.[9a] This concept of hydrogen bonding was also assumed in the L-proline (14) catalyzed Mannich reactions.[69] A better soluble alternative to L-proline (14) in Mannich reactions is the tetrazole catalyst 32.[22, 27f] Furthermore, Diels-Alder reactions[70] as well as hetero Diels-Alder reactions[71] are known to be catalyzed over hydrogen-bonding interactions with the TADDOL-derived catalyst 70.
Figure 5. Brønsted acids acting as organocatalysts in different reactions.
The quinidine catalyst 71 was found to catalyze Baylis-Hillman[72] as well as aza-Baylis Hillman reactions[73] in excellent yield and stereoselectivity, which is referred to the H-bonding ability of the hydroxy group. Hiemstra et al. discovered that the cinchona alkaloid derivative 72 served as outstanding catalyst for the enantioselective Henry reaction.[74]
Cinchona alkaloid 73 was used in a Friedel-Crafts addition of indole to ethyl trifluoropyruvate and thereby the necessity of the hydroxyl group for the formation of hydrogen-bonding was proofed.[75] Moreover, catalyst 73[76] and the tertiary amine-thiourea derivative 74[77] were both utilized in the Michael addition.
These examples reflecting only a small section of the great diversity of H-bond donor catalysts developed during the last years.
5. Brønsted Base Catalysis
The Brønsted base catalysis is a hardly classifiable field, due to the fact that many organocatalyst are bifunctional. Thus, an urea catalyst that bears an amine could be seen either as Brønsted base catalyst or as an H-bonding donor catalyst.[2] Typical examples of Brønsted base catalysts are displayed in Figure 6.
Figure 6. Organic Brønsted bases for organocatalysis.
Typical reactions in this field are hydrocyanation reactions like the cyanohydrin synthesis and the Strecker reaction.[3] The cyclopeptide 75, for example, was used in a HCN addition reaction to various aldehydes.[78] Moreover, Corey and Grogan studied the Strecker reaction using a chiral C2-symmetric guanidine 76.[79] Furthermore, Isobe et al. reported a Michael reaction of a prochiral glycine derivative catalyzed by the modified guanidine 77.[80] Deng and co-workers applied the modified cinchona alkaloid 78 as catalyst to the desymmetrization of cyclic meso-anhydrides.[81] The latter mentioned desymmetrization of meso compounds has developed to a powerful tool in asymmetric synthesis and especially in Brønsted base organocatalysis in the last few years (for a detailed review see reference [82]). In conclusion, these different examples make clear that Brønsted base catalysts is a fast growing and important sector of organocatalysis.
6. Lewis Acid Catalysis
Lewis acid catalysis is a small and often neglected but important activation mode in organocatalysis. The main focus of research was laid on Lewis base (enamine/iminium catalysis) and Brønsted acid catalysis although it can develop to an equal powerful tool.[83] By taking a closer look, it gets obvious that a big part of Lewis acid organocatalysts can also be seen as phase-transfer catalysts[3] and often these catalysts bear a positively charged center that is prone to activate the substrate.[84] Figure 7 displays a few examples of Lewis acid organocatalysts.
Figure 7.Examples of Lewis acid organocatalysts.
Wilhelm et al. used the bis-imidazolinium salt 79 in a Diels-Alder reaction and obtained the product in excellent endo:exo-selectivity with good yield. Moreover, they applied catalyst 79 and 80 in the ring opening reaction of cyclohexene epoxide with outstanding results.[84] Aryl cinchoninium salt 81 was one of the first examples of an Lewis acid organocatalyst developed by Dolling and co-workers in 1984.[85] It was applied to an -alkylation of indanone under phase-transfer catalytic conditions. Another example that shows a great applicability of Lewis acid organocatalysts to various C-C-bond forming reactions is the C2-symmetric chiral spiro
ammonium salt catalyst 84. It was utilized to -alkylations[86], aldol[87] and Michael reactions[88]. On the other hand, also uncharged molecules are known that serve as Lewis acid catalysts. One important class are the in situ formed chiral dioxiranes generated from chiral ketone catalysts and Oxone (potassium peroxomonosulfate).[89] Shi et al. used the D-fructose derived ketone catalyst 82 for an enantioselective epoxidation of various olefines.[90]
Furthermore, the imide 83 was found to be an excellent catalyst for the Friedel-Crafts alkylation[91] and different cycloaddition reactions[92]. The BINOL-derived disulfonimide 85 was developed by List and co-workers and was applied to the Mukaiyama aldol reaction to obtain outstanding results.[83] These manifold examples show the great potential of Lewis acid catalysis especially in the field of phase-transfer catalysis.
All in all, the literature examples presented above demonstrate the broad applicability of organocatalysts to many reactions. Moreover, it seems obvious that from such an easy modifiable and robust class of catalysts creative ideas for developing new and powerful catalysts could arise in the future. In the present thesis, attempts for the optimization and investigation of an L-proline/Co(II)-catalyzed aldol reaction, as well as the subsequent application of this complex to other reactions were done. Moreover, easy accessible 1,2-diamio alcohols were utilized to various organic transformations to show their broad applicability. The results are presented in the following.
B Organocatalysis of L-Proline in the Presence of Metal Salts 1. Introduction
1.1 The Nonlinear Effect (NLE) - From the Discovery to Present Research
An important aim for organic chemists has always been the development of reactions and strategies that induce a high level of stereoselectivity in the desired target molecule.[93] The discovery of chirality dates back to the year 1848, when Luis Pasteur examined the ammonium sodium salt of tartaric acid and thereby recognized that it rotates the plane of polarized light in two different ways depending on its origin.[93b, 94] Moreover, he realized that a racemic mixture of compounds has no effect on the rotation of polarized light and is therefore optical inactive. Thus, a linear correlation between the enantiomeric excess (ee) and the optical purity was concluded making the use of polarimetry for determining the ee the method of choice. Nevertheless, further investigations lead to the finding of a deviation of this linear correlation, which was explained by the nature and composition of the present mixture and its enantiomeric purity, as pure enantiomers compared to racemic mixtures can have a different chemical rate and thus generating a different product distribution. The pioneering work in this field was done by Horeau et al.[95] who were the first to notice this phenomenon of the not exact linear correlation between specific rotation and enantiomeric excess.[94b, 94c, 96]
Further work in this area was done by Wynberg and Feringa[97], who examined the non-ideal behavior of a mixture of enantiomers in solution and pointed out that diastereoselective reactions are influenced by the ee of the substrate. The reason for this is that in a racemic mixture interactions between the two enantiomers could develop that are not possible in an enantiopure system. As a consequence these mixtures differ in their influence on reactivity and stereoinduction.[93b, 94b, 94c, 96]
In 1986 Kagan et al. explained, for the first time, the quantitative aspects of the nonlinear correlation between the ee of the auxiliary (eeaux) and the ee of the product (eeprod), proved by three examples from asymmetric catalysis (Scheme 7).[93b, 93c, 94b, 94c, 96, 98]
They coined it “nonlinear effect” (NLE), whereas over time and further investigations terms like chiral/asymmetric amplification, chiral multiplication or asymmetric depletion occurred.[93b, 93c, 96, 98b, 99]
Scheme 7. Examples of the first NLEs in literature; a) (−)-NLE in the Hajos-Parrish-Wiechert reaction of 86 to 87 catalyzed by (L)-proline (14); b) (+)-NLE in the Sharpless epoxidation of geraniol (88); c) (−)-NLE until 70%
(from then on linearity was resumed) in the asymmetric oxidation of sulfide 90 with a chiral titanium reagent.[93b, 93c, 94b, 94c, 96, 98]
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
a)
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
b)
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
c)
Diagram a) in Scheme 7 shows the intramolecular asymmetric aldolization of triketone 86 catalyzed by L-proline (14) for which a small negative nonlinear effect [(-)-NLE)] was assumed. However, more recent studies by List et al. disproved this assumption.[100] A positive nonlinear effect [(+)-NLE] was noticed in the Sharpless epoxidation of geraniol (88) [diagram b), Scheme 7]. Furthermore, the asymmetric sulfoxidation of 90 generated a (-)-NLE, until 70% ee were reached, from then on linearity was resumed till a maximum of 85% ee [diagram c), Scheme 7].[94b, 98a]
The correlation of the ee value of the product in an asymmetric reaction with the ee value of the chiral auxiliary could easily be done if the latter are acting independently from each other.
For this case equation (1) can be applied in which the ee of the product correlates in a linear way with the ee of the auxiliary. The maximum ee value of the product (eemax) is obtained by carrying out the catalysis with the enantiopure chiral catalyst or auxiliary.[93c, 94b, 96a]
𝑒𝑒𝑝𝑟𝑜𝑑(%) = (𝑒𝑒𝑚𝑎𝑥× 𝑒𝑒𝑎𝑢𝑥) × 100 (1)
After transforming equation (1) into equation (2), it is possible to specify eeprod by using the linear relationship given in equation (2)
𝑒𝑒𝑚𝑎𝑥(%) = (𝑒𝑒𝑝𝑟𝑜𝑑÷ 𝑒𝑒𝑎𝑢𝑥) × 100 (2)
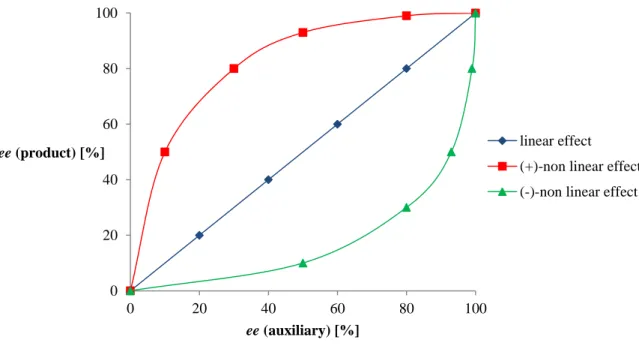
If eeprod is plotted versus eeaux as displayed in Figure 8 in the case described above a linear correlation should occur (blue line, Figure 8). However, there also might occurr a positive nonlinear effect (red line, Figure 8) or a negative nonlinear effect (green line, Figure 8). It should be mentioned that the curves in Figure 8 correspond to an ideal case and are not generalizable, since there are known literature examples of a combination of linear and nonlinear effects, e.g. in the asymmetric sulfoxidation described above (Scheme 7).[98a]
Figure 8General graph displaying the three cases of a linear (blue line) correlation and a positive (red line) or a negative (green line) nonlinear correlation between eeprod and eeaux.
This nonlinear phenomenon can appear, if the catalyst contains two or more chiral ligands that are not enantiomerically pure, because in this scenario homochiral or heterochiral catalytic species are formed, which differ in their reaction rate and stereoinduction. Therefore, the linear relationship expressed in equation (1) and (2) is no longer valid, requiring their adjustment by introducing correction factors. Many theoretical models to describe such non- linear effects were established in the last two decades. The simplest situation is encountered when two enantiomeric chiral ligands (LR and LS) are attached to a metal center (M) to generate ML2 complexes as reactive species. Owing to this situation it is possible to get three different species, two homochiral ones [M(LR)2 and M(LS)2] and one hetero- or mesochiral one (MLRLS). Scheme 8 illustrates the case for a dynamic equilibrium between the three complexes M(LR)2, M(LS)2 and MLRLS and a fast exchange of the ligands (LR and LS) at the metal (M).
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
linear effect (+)-non linear effect (-)-non linear effect
Scheme 8. Overview of the ML2 model system.[101]
In this model system the two homochiral complexes are forming the two opposite enantiomeric products, whereas the heterochiral species is forming exclusively the racemic product with respect to their relative reactivity (g = kRS/kRR or kRS/kSS) and their relative concentrations [ = z/(x+y)][93c, 102] (Scheme 8).[101] With these assumptions, it is now possible to formulate equation (3), which expresses the eeprod as a function of eeaux (red and green curve, Figure 8) [101]
𝑒𝑒𝑝𝑟𝑜𝑑(%) = [(𝑒𝑒𝑚𝑎𝑥× 𝑒𝑒𝑎𝑢𝑥) ×1+𝑔𝛽1+𝛽] × 100 (3)
The values for g and eemax are fixed for a certain system and can be calculated with the help of the equilibrium constant K between the heterochiral and homochiral complexes.[101] The requirement for equation (3) to be valid is that all ligand (LR and LS) is converted into one of the three metal complexes [M(LR)2, M(LS)2 or MLRLS] or that the external ligand is enantiopure and therefore maintains the initial value of eeaux. Thus, three different cases may occur. When = 0 (no meso catalyst is forming) or g = 1 (reactivities of meso and homochiral catalysts are identical) equation (3) is simplified to equation (2) and a linear correlation between eeprod and eeaux (blue curve, Figure 8, page 19) is obtained. When g > 1 a negative nonlinear effect is observed which at the same time means that the meso complex is more reactive than the homochiral one (green curve, Figure 8, page 19). If g < 1 the reversed case is true. The homochiral complex is more reactive compared to the meso complex, hence leading to a positive nonlinear effect (red curve, Figure 8, page 19). As one might expect, the biggest deviation from linearity in a positive NLE is obtained when g = 0 meaning that the meso complex is catalytic inactive.[96a] By reaching the thermodynamic equilibrium, the highest positive NLEs can be achieved if the equilibrium constant K is large and g is small which, in other words, means the reaction takes place very slowly and the concentration of the meso
complex is high.[96a] Blackmond et al. investigated the NLE with regard to kinetic aspects, because this point of view was often neglected by other groups.[96b] Based on the assumption that the value of K is fixed, there are two cases for non-linearity (summarized in Scheme 9).
When an asymmetric amplification [(+)-NLE] is present the reaction rate is decreases if the eeaux decreases. In contrast, if there is an asymmetric depletion [(−)-NLE] the reaction rate increases if the eeaux decreases. Therefore, it is necessary to decide whether the product should be formed in a big amount but with low ee value (high reactivity) or in small quantities but with high enantiomeric purity (low reactivity).[96b, 98b]
Scheme 9. Relations between reaction rates and the presence or absence of nonlinear effects.[98b]
For systems with more than two ligands (ML3, ML4...MLn,) or for the, so-called reservoir effect, in which the catalytic active species is partially transferred into a catalytic inactive species also theoretical models were investigated, but in the course of this work it is not further focused on these models. For more detailed information of these models see the publications of Kagan et al..[93b, 94b, 98b, 101]
Since the discovery and the detailed investigation of the NLE by Kagan et al. many reactions catalyzed by a combination of a metal and two or more chiral ligands were examined under the focus of asymmetric amplification. One important example in homogeneous asymmetric catalysis is the asymmetric addition of organozinc compounds to aldehydes, which was intensively analyzed under the aspects of this effect by many working groups, inter alia Oguni et al.[93c, 103]
, Noyori et al.[93c, 102, 104]
, Bolm et al.[93c, 105]
and also Kellog et al.[93c, 106]
. Scheme 10 shows the asymmetric addition of diethylzinc (93) to benzaldehyde (92) catalyzed by different ligands 95-103 which show either a (+)-NLE or a (−)-NLE.[93c, 102-106]
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
a)
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
catalyst 96 catalyst 97 catalyst 98
b)
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
c)
Scheme 10. First examples of asymmetric amplifications in the asymmetric addition of Et2Zn (93) to benzaldehyde (92); the graphs on the left side show the correlation between the ee of product (94) and the ee of the auxiliary, investigated for the catalyst a) 95 by Noyori et al.[104a]; b) 96-98 by Oguni et al.[103]; c) 99 by Bolm et al.[105]; d) 100-103 by Kellog et al.[106].
In the course of the years, many other homogeneous organometallic catalyzed reactions with NLEs were found, e.g. conjugated additions of organometal compounds to enones, allylation of aldehydes, cyanide addition to carbonyl groups, C-alkylations, epoxide openings and rearrangements, enantioselective oxidations, reductions and Diels-Alder reactions.[93c]
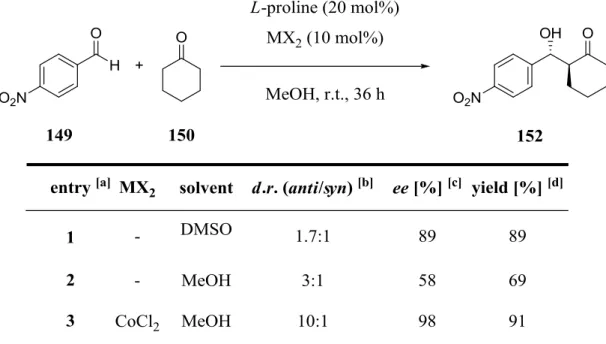
Nevertheless, here no closer look is taken on these reactions because they did not deal with the reactions which are investigated in this work. For an overview of these reactions see the review of Kagan et al..[93c] Two, so far not mentioned homogeneous organometallic catalyzed reactions are aldol and Mannich reactions, respectively. As part of this work the NLE for the direct asymmetric aldol reaction between cyclohexanone (150) and p-nitrobenzaldehyde (149) catalyzed by a combination of L-proline (14) and CoCl2 was analyzed. Therefore, some examples of aldol additions with respect to asymmetric amplification are presented in detail on the next few pages.
Another big field besides homogeneous organometallic catalysis is represented by homogeneous organocatalysis, which was intensively investigated in terms of NLEs. As mentioned before, the Robinson Annulation was the first reaction in this field which showed a NLE and was investigated by Kagan, Agami and co-workers (Scheme 7, page 17).[98a]
However, List and co-workers reexamined the reaction and observed no deviation from linearity between the ee of the auxiliary and the ee of the product. Therefore, they made the assumption that only one L-proline (14) molecule is involved in the transition state of the
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
catalyst 100 catalyst 101 catalyst 102
d)
examples of an asymmetric amplification in L-proline (14) catalyzed reactions are shown in Scheme 11.
Scheme 11. The only two literature known L-proline (14) catalyzed reactions showing an asymmetric amplification; the graphs on the left side show the correlation between the ee of the product and the ee of the auxiliary; a) self aldol addition of propionaldehyde (104) [Eq. (1)]; b) Mannich type reaction between propionaldehyde (104) and N-protected amino glyoxylate 106 [Eq. (2)].[93c, 107]
Equation (1) in Scheme 11 shows the asymmetric formation of the aldol addition product 105 catalyzed by L-proline (14), whereby a significant asymmetric amplification was noticed by Cordova et al..[107a] Since only one L-proline (14) molecule is involved in the catalytic process, the (+)-NLE must be caused by the product 105 itself. It is hypothesized that the amplification is based on the fact of different rates of reactivity of L- (14) or R-proline (110) with the sugar precursor 105. Therefore, the latter is “auto”-kinetically resolved leading to an enrichment of the free amino acid in the next catalytic cycle and hence to a positive nonlinear effect (Scheme 12).[93c, 107a]
Equation (2) in Scheme 11 displays the Mannich type reaction of propionaldehyde (104) with the N-protected -amino glyoxylate 106. The studies of
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
a)
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
b)
Cordova et al. on this reaction also revealed an asymmetric amplification. Based on earlier mechanistic studies on Mannich type reactions it was supposed that also here only one proline molecule is involved in the formation of amino acid derivative 107.[15b, 108]
Based on these facts it is proposed that the amino acid 107 is probably reacting at different rates with the L- (14) or R-proline (110) and therefore “auto”-kinetically resolves the amino acid catalyst by forming oxazolidine structures which are leading to an (+)-NLE in the next catalytic cycle.[93c, 107b]
This is in accordance with the explanation given for the aldol reaction of propionaldehyde (104) (see Eq. (1), Scheme 11 and Scheme 12).[107b]
Scheme 12. Different reaction rates of L- (14) and R-proline (110) with sugar 105.[93c, 107a]
Close related with these findings is the discovery of a positive nonlinear effect in the neogenesis of carbonhydrates catalyzed by L-proline (14) or 4-hydroxy-L-proline (114), respectively, by Cordova et al. (Scheme 13).[109] The reaction proceeds in the following way:
First a self aldol addition takes place forming erythrose 116, followed by the addition of erythrose 116 to a second enamine 115 leading to the allose intermediate 117, which subsequently is transformed to the more stable hexose 118. The latter step is also the rate-determing one. Hence, the interaction between non-enantiomerically pure proline and the tetrose 116 was given as explanation for the asymmetric amplification in the aldol addition.[109]
Scheme 13. L-proline (14) catalyzed asymmetric one-step synthesis of allose 118 and the observed NLE.[109]
Blackmond et al. examined the two reactions shown in Scheme 14, the L-proline (14) catalyzed a-aminoxylation [Eq. (1)][110] and the -amination [(Eq. (2)][111] of propionaldehyde (104).
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
Scheme 14. The graphs on the left side show the correlation between the ee of the product and the ee of the auxiliary; a) asymmetric amplification of the L-proline (14) catalyzed -aminoxylation of propionaldehyde (104) [Eq. (1)]; b) asymmetric amplification of the L-proline (14) catalyzed -amination of propionealdehyde (104) [Eq. (2)].[110-111]
In both reactions an abnormal increase in the reaction rate and (+)-NLE was found probably, due to the formation of a proline adduct with product 120 or 122, which in the further course of the reaction acts as a superior catalyst compared to L-proline (14). Thus, the asymmetric amplification is explained by kinetic resolution of proline by the reaction with product 120 or 122, respectively, which can be seen as a selectivity-enhancing autoinductive process.[110-111]
In conclusion these are the only examples where L-proline (14) shows a nonlinear effect.
However, one further case is described in literature where a catalyst 118 similar to L-proline (14) is leading to asymmetric amplification, which was discovered by Jørgensen and co-workers (Scheme 15).[93c, 112]
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
a)
0 20 40 60 80 100
0 20 40 60 80 100
ee (product) [%]
ee (auxiliary) [%]
b)