AUS DER KLINIK FÜR ANÄSTHESIOLOGIE Direktor: Prof. Dr. med. Bernhard M. Graf, MSc.
DER FAKULTÄT FÜR MEDIZIN DER UNIVERSITÄT REGENSBURG
CHOLINERGE IMMUNMODULATION HUMANER NEUTROPHILER GRANULOZYTEN
Inaugural Dissertation zur Erlangung des Doktorgrades
der Medizin
Fakultät für Medizinder der Universität Regensburg
vorgelegt von Christoph Paech
2015
AUS DER KLINIK FÜR ANÄSTHESIOLOGIE Direktor: Prof. Dr. med. Bernhard M. Graf, MSc.
DER FAKULTÄT FÜR MEDIZIN DER UNIVERSITÄT REGENSBURG
CHOLINERGE IMMUNMODULATION HUMANER NEUTROPHILER GRANULOZYTEN
Inaugural Dissertation zur Erlangung des Doktorgrades
der Medizin
Fakultät für Medizinder der Universität Regensburg
vorgelegt von Christoph Paech
2015
Dekan: Prof. Dr. Dr. Torsten E. Reichert 1. Berichterstatter: PD Dr. Benedikt Trabold 2. Berichterstatter: PD Dr. Bernhard Flörchinger Tag der mündlichen Prüfung: 02. Dezember 2015
Diese Arbeit widme ich meinen Eltern als Dank für ihre stete Unterstützung.
Inhaltsverzeichnis Inhaltsverzeichnis
1 Einleitung ... 1
1.1Sepsis und Inflammation... 1
1.2 Neutrophile Granulozyten und Endothel ... 1
1.2.1 Selektine... 3
1.2.2 Selektin-Liganden ... 4
1.2.3 Integrine ... 4
1.2.4 Immunglobulin-Superfamilie... 5
1.2.5 Mehrstufenmodell der Leukozytenadhäsion und -migration... 6
1.3 Sauerstoffradikalbildung... 9
1.4 Die Proteinkinase C... 10
1.5 Das non-neuronale cholinerge System... 11
1.6 Fragestellung ... 14
2 Material und Methoden... 16
2.1 Material ... 16
2.1.1 Geräte ... 16
2.1.2 Verbrauchsmaterial ... 17
2.1.3 Reagenzien ... 17
2.1.4 Puffer-Lösungen... 19
2.2 Methoden... 19
2.2.1 Probanden... 19
2.2.2 Getestete Substanzen und Konzentrationen... 20
2.2.3 Isolation der neutrophilen Granulozyten ... 21
2.2.3.1 Durchflusszytometrie... 21
2.2.3.2 ELISA ... 21
2.2.4 Messung der Sauerstoffradikalbildung ... 22
2.2.4.1 Theoretische Grundlagen der Messung ... 22
2.2.4.2 Ablauf der Messung ... 22
2.2.5 Messung der CD11b- und CD62l-Expression ... 24
Inhaltsverzeichnis
2.2.6 Durchflusszytometrie (FACS-Messung) ... 24
2.2.7 Quantifizierung der Proteinkinase C - Aktivität... 26
2.2.7.1 Der ELISA ... 26
2.2.7.2 Bestimmung der Proteinkonzentration ... 28
2.2.7.3 Probandenproteine ... 28
2.2.7.4 Gereinigte Proteinkinase C - delta ... 29
2.2.8 Versuche mit HUVEC ... 29
2.2.8.1 An- und Aufzucht... 29
2.2.8.2 Versuchsdurchführung mit Acetylcholinchlorid ... 32
2.2.9 Statistische Analyse ... 33
3 Ergebnisse ... 34
3.1 Durchflusszytometrie... 34
3.1.1 Physostigminsalicylat ... 34
3.1.2 Acetylcholinchlorid... 36
3.1.3 Nikotin ... 37
3.1.4 Anteil toter Zellen... 42
3.1.5 Acetylcholinwirkung auf die HUVEC-Antigenexpression ... 43
3.2 Proteinkinase C - ELISA ... 44
3.2.1 Probandenproteine ... 44
3.2.2 Gereinigte Proteinkinase C - delta... 45
4 Diskussion... 46
4.1 Durchflusszytometrie... 46
4.1.1 Physostigminsalicylat ... 46
4.1.2 Nikotin ... 47
4.1.3 Acetylcholinchlorid... 50
4.1.3.1 Neutrophile Granulozyten... 50
4.1.3.2 HUVECs ... 51
4.2 Proteinkinase C-ELISA ... 53
4.3 Schlussfolgerungen für die zelluläre Ebene des cholinergic antiinflammatory pathway ... 55
Inhaltsverzeichnis
5 Zusammenfassung... 58
6 Literaturverzeichnis... 60
7 Abbildungsverzeichnis... 70
8 Tabellenverzeichnis... 72
9 Abkürzungsverzeichnis... 73
10 Danksagung... 75 0
Anhang ...
Erklärung zum Promotionsverfahren...I
Einleitung
1 Einleitung
1.1 Sepsis und Inflammation
Jeder Mensch ist tagtäglich einer Vielzahl verschiedenster Noxen ausgesetzt.
Nebst effektiven Schutzbarrieren wie der Haut, ist ein funktionierendes und universell agierendes Immunsystem daher unabdingbar. Auf der einen Seite müssen effiziente Abwehrstrategien vorhanden sein, auf der anderen Seite dürfen selbige nicht den eigenen Organismus schädigen. Wie der menschliche Körper diese Balance regelt, ist Gegenstand intensiver Forschung. Fest steht, dass eine überschießende Immunreaktion des Organismus fatale Folgen haben kann.
Die systemische Inflammation stellt trotz aller hochtechnologisierter Medizin noch immer eine große Herausforderung in der intensivmedizinischen Betreuung dar. Dabei zählt die infektbedingte systemische Inflammation (Sepsis) zu den zehnthäufigsten Todesursachen in den USA und setzt sich somit noch vor schlaganfall- und krebsassoziierte Todesfälle.1
1.2 Neutrophile Granulozyten und Endothel
Gewebeschäden jedweder Genese (Trauma, Infektionen etc.) bewirken ein komplexes Zusammenspiel aus immunologischen Effektorzellen, Endothel und umliegendem Gewebe. Eine Schlüsselrolle bei inflammatorischen Prozessen spielen die neutrophilen Granulozyten.2
Als neutrophile Granulozyten wird eine Subpopulation leukozytärer Blutzellen bezeichnet. Dabei handelt es sich um phagozytäre Zellen der angeborenen Immunabwehr, die mit 50% 65% den größten Anteil der weißen Blutkörperchen bilden. Sie treten aus der Blutbahn in das geschädigte Gewebe über und entfalten dort ihre immunologische Funktion.
Die Leukozyten-Endothel-Adhäsion stellt einen frühen und geschwindigkeitsbestimmenden Schritt der Leukozyten-Infiltration dar. Hierbei
Einleitung
bewirken zeitlich sowie örtlich genau abgestimmte physikalische und chemische Mediatoren eine Herauf- oder Herunterregulation der Immunantwort. Zunächst kommt es zu einem Entlangrollen der Neutrophilen auf der Endotheloberfläche, dies wird durch schwache Adhäsionskräfte ermöglicht. Sobald der Leukozyt ausreichend aktiviert ist, stellt sich eine feste Adhäsionsbindung zwischen ihm und dem Endothel ein. Kurz darauf erfolgt die Diapedese in subendotheliales Gewebe.
Im Folgenden sollen die relevanten Familien der Adhäsionsmoleküle sowie deren wichtigste Vertreter kurz vorgestellt (s. Tabelle 1) und anschließend detaillierter auf das Mehrstufen-modell des Zellübertritts vom Blutstrom in das umgebende Gewebe eingegangen werden.
Adhäsions-
molekül Zellart Expression
Ligand Funktion Ruhe-
zustand Induzierbar Selektine L-Selektin
(CD62l) Alle
Leukozyten Zellaktivierung:
Expression P-,E-Selektin
CD34 Rollen
P-Selektin Endothel,
Thrombozyten L-Selektin Rollen
E-Selektin Endothel Nein L-Selektin Rollen
Integrine CD11a/CD18 Alle
Leukozyten Nein ICAM-1,-2 Adhäsion, Migration CD11b/CD18 Granulozyten, Monozyten ICAM-1, C3b Adhäsion, Migration CD11c/CD18 Granulozyten,Monozyten ICAM-1,
Fibrin Adhäsion, Migration
Einleitung
Immunglobulin Superfamilie
ICAM-1 Endothel LFA-1, Mac-1 Adhäsion,
Migration
ICAM-2 Endothel Nein LFA-1 Adhäsion,
Migration
VCAM-1 Endothel Nein VLA-4 Adhäsion,
Migration PECAM-1 Endothel,
Leukozyten,
Thrombozyten Nein PECAM-1 Adhäsion,
Migration Tabelle 1 Übersicht der wichtigsten humanen Adhäsionsmoleküle19122
1.2.1 Selektine
Die Gruppe der Selektine wird unterteilt in L-, E- und P-Selektine. Diese Bezeichnungen ergeben sich aus ihrer vorrangigen Präsenz auf Leukozyten (L- Selektine), Endothelzellen (E-Selektine) sowie Blutplättchen und Endothelzellen (P-Selektine).3Selektine gehören chemisch betrachtet zu den Glycoproteinen und sind ausschließlich im Gefäßsystem funktionell aktiv.
Monozyten, neutrophile und eosinophile Granulozyten sind in der Regel L- Selektin-positiv. Die L-Selektine spielen hier bei der Bindung von den entsprechenden Immunzellen an aktivierte Endothelzellen eine Rolle. An unstimulierten Gefäßwänden konnte dieser Effekt hingegen nicht beobachtet werden, was wiederum als ein Indiz für die fehlende Expression des entsprechenden Liganden an ruhenden Gefäßzellen zu werten ist.4Leukozyten benötigen selbst keinen aktivierenden Stimulus für die selektinvermittelte Interaktion mit dem Endothel. Im Gegenteil, eine Stimulation führt zu einer Reduktion der auf der Plasmamembran exprimierten L-Selektine.5,6
P-Selektine finden sich, wie schon erwähnt, gleichermaßen am Endothel, wie auch an Blutplättchen und fungieren als Rezeptoren für Makrophagen und neutrophile Granulozyten.7,8Auf Endothel exprimierte P-Selektine dienen der
Einleitung
Rekrutierung von Leukozyten zu entsprechenden Entzündungsherden.9Zudem wurde auch die Initiierung der Sauerstoffradikalbildung durch Neutrophile und Monozyten an aktivierten Thrombozyten beobachtet, während dies an den ruhenden Thrombozyten sowie nach Gabe gegen P-Selektin gerichteter Antikörper nicht der Fall war.10Die synthetisierten P-Selektine werden in den Weibel-Palade-Körperchen und Alpha-Granula der entsprechenden Immunzellen gespeichert.7Diese Speicher werden innerhalb weniger Minuten nach Aktivierung durch Histamin, Komplementfaktor C5q oder Superoxidanionen in die Plasmamembran integriert und die darin enthaltenen Adhäsionsmoleküle auf der Zelloberfläche präsentiert.11 Aufgrund von Internalisierung und Abstoßung verringert sich die P-Selektin-Dichte nach 30 Minuten wieder zurück auf das Ausgangsniveau.12 Des Weiteren findet auch eine transkriptionsgebundene Expression von P-Selektin statt. Dies wurde beobachtet, nachdem Endothelzellen vier Stunden zuvor mit bakteriellen Lipopolysacchariden beimpft wurden.13 Die dritte Gruppe der Selektinfamilie, die E-Selektine, sind physiologischerweise nicht auf unstimulierten Endothelzellen zu finden. Ihre Expression findet mittels induzierter Transkription nach Stimulation mit Zytokinen statt.14Hierbei zeigte sich im Mausmodell ein Expressionspeak nach zwei sowie eine Normalisierung nach acht Stunden.15,16
1.2.2 Selektin-Liganden
Der am weitesten verbreitete L-Selektin-Ligand im Blutgefäßsystem ist CD34. Er dient als Ligand für Lymphozyten und Neutrophile in der Peripherie.2
Ähnlich wie bei E- und P-Selektin steht die Expression von CD34 unter Zytokineinfluss. Jedoch bewirken Interferon- , Interleukin-1 und Tumornekrosefaktor-alpha (TNF- ) bei Konzentrationen, die positiv auf die endothelexprimierten Selektine E und P wirken, eine herunterregulierte CD34- Expression. Dies erscheint zunächst unlogisch. Doch die Selektine weisen auch untereinander Bindungseigenschaften auf. So wirkt L-Selektin sowohl als
Einleitung
Bindungspartner für E-, als auch P-Selektin und vice versa.17,18 Die nach Stimulation vermehrt exprimierten Selektine des Endothels erzeugen in der Interaktion mit den L-Selektinen der Immunzellen ausreichend starke Bindungskräfte, um die nachfolgenden Schritte der Leukozyten-Endothel- Interaktion zu ermöglichen.
1.2.3 Integrine
Einen weiteren wichtigen Baustein in der Leukozyten-Endothel- Adhäsionskaskade bilden die Integrine. Dabei handelt es sich um heterodimere Proteine, die aus zwei nicht-kovalent gebundenen Untereinheiten bestehen, wobei
d sind. Leukozyten exprimieren unter
anderem fünf Integrine. Diese -Subtypen zugeordnet und gehören somit zu den an der Leukozyten-Endothel-Interaktion beteiligten Untergruppen.19
Fü 1- und 2-Integrine ist insbesondere die
variierende alpha-Untereinheit von Bedeutung.19 -Grundbaustein (CD18) tritt im Komplex mit drei immunologisch unterscheidbaren alpha-Ketten auf:
CD11a, CD11b und CD11c. Das Expressionsmuster unterscheidet sich bei den einzelnen Leukozytenarten, wobei die neutrophilen Granulozyten jedoch alle drei
-Subtypen ausprägen.19
CD11a/CD18 ist konstitutiv exprimiert, also auch an immunologisch unstimulierten Zellen und interagiert mit endothelialem intercellular adhesion molecule (ICAM)-1 und -2.20 Dieser Integrin-Subtyp verfügt nicht über eine intrazelluläre Reserve in Form von Speichergranula, welche nach Stimulation in die leukozytäre Plasmamembran integriert und somit zu einer Zunahme der Leukozyten-Endothel-Adhäsion führen würde. Dennoch bewirkt eine Stimulation mit Formyl-Methionyl-Leucyl-Phenylalanin (fMLP) oder Interleukin-8 eine rapide Adhäsionszunahme der Leukozyten am Endothel, welche am ehesten durch eine konformative Veränderung der Integrin-Affinität zu erklären ist.21
Einleitung
Demgegenüber stehen CD11b/CD18 und CD11c/CD18. Sie besitzen eine intrazelluläre Reserve, die nach Stimulation zur Verfügung steht.22 Die Expressionsdichte kann nach Stimulation mit TNF-alpha oder anderen Stimuli auf das Drei- bis Achtfache ansteigen.23 CD11b/CD18 interagiert ebenfalls mit ICAM-1, während CD11c/CD18 zusätzlich an Heparin und Komplementfaktoren bindet.24,25,26
Die Adhäsion aktivierter neutrophiler Granulozyten an zytokinstimuliertem Endothel lässt sich sowohl durch anti-CD11a, als auch durch anti-CD11b Antikörper verhindern. Hierbei bewirkt die simultane Gabe beider Antikörper einen additiven Hemmeffekt.27Diese Beobachtungen unterstreichen die Relevanz der Integrine im immunologischen Abwehrprozess.
1.2.4 Immunglobulin-Superfamilie
Als drittes und letztes Adhäsionsmolekül möchte ich kurz auf die Immunglobulin- Superfamilie eingehen. Diese besteht aus mehreren Vertretern, denen alle eine Immunglobulin-ähnliche Domäne gemein ist. Für die Leukozyten-Endothel- Interaktion sind dabei die folgenden fünf Subtypen relevant, von denen zuvor schon einige genannt wurden: ICAM-1 (CD54), ICAM-2 (CD102), VCAM-1 (vascular cell adhesion molecule, CD106), PECAM-1 (plateled-endothelial cell adhesion molecule, CD31) und MAdCAM-1 (mucosal adressin cell adhesion molecule).
Da sich unser Projekt auf neutrophile Granulozyten bezieht, beschränken sich die anschließenden Erläuterungen auf ICAM-1 und VCAM-1 den getesteten Parametern. Schon im Ruhezustand wird ICAM1 auf Leukozyten, Endothelzellen und anderen Zellarten exprimiert. Hierbei kann die Rezeptordichte auf Endothelzellen durch deren Aktivierung via LPS oder Zytokine erhöht werden.13,28Sowohl die Funktion als auch die physiologische Relevanz dieses Adhäsionsmoleküls erklärt sich durch die Beobachtung, dass eine
Einleitung
erhöhte Rezeptordichte mit einer signifikant höheren Leukozytenadhäsion am Endothel einhergeht.29
Ähnlich verhält es sich mit dem vascular cell adhesion molecule 1(VCAM- 1). Hierbei handelt es sich um ein Endothelzell-spezifisches Adhäsionsmolekül, welches auf unstimulierten HUVECs (human umbilical-vein endothelial cell) nicht nachweisbar ist. Erst durch Stimulation mit LPS oder Zytokinen erfolgt die transkriptionsabhängige Expression.30,31Daher ist der Expressionspeak auch erst fünf bis neun Stunden nach Zytokin-Stimulation nachweisbar und liegt dabei noch deutlich unter der Zelloberflächendichte von ICAM-1.32 VCAM-1 ist ein wichtiger Modulator der Leukozytenadhäsion und dient als Ligand sowohl für
4 1-Integrin33 4 7 -Integrin34, wobei die letztgenannte Bindung weniger stark ausfällt. Dementsprechend ist es nicht verwunderlich, dass zahlreiche Faktoren auf die Oberflächendichte dieser Immunglobulin- Superfamilie Einfluss nehmen.
So können LPS sowie die - -4 und
Interferon- die Expression von P- und E-Selektin, als auch von VCAM-1 und ICAM-1 signifikant erhöhen.13,35,36 Auf HUVEC-Monozellschichten konnte hierbei beobachtet werden, dass die Zelloberflächendichte von P- und E-Selektin drei bis fünf Stunden, von VCAM-1 sechs Stunden und von ICAM-1 zwölf Stunden nach Stimulation am höchsten ist.37,38,39
1.2.5 Mehrstufenmodell der Leukozytenadhäsion und -migration
In den vorangegangenen Abschnitten wurden die einzelnen molekularen Faktoren, welche für die Leukozyten-Endothel-Interaktion relevant sind, kurz vorgestellt. Im Folgenden soll nun deren komplexes Zusammenspiel anhand des Mehrstufenmodells erläutert werden. Denn nur durch das chronologisch aufeinander abgestimmte Zusammenwirken aller Gruppen, können der Übertritt der immunologischen Effektorzellen aus dem Gefäßsystem in das erkrankte Gewebe erfolgen und die effektiven Abwehrmechanismen greifen.
Einleitung
Um überhaupt eine Adhäsion der Leukozyten mit der Gefäßwand zu erreichen, müssen diese in einem initialen Schritt vom Zentralstrom des Blutes in die langsameren peripheren Strömungen der Blutgefäßwände abgelenkt werden. Dies geschieht während des Übertritts von den kleineren Kapillaren zu den größeren postkapillaren Venolen.40Hierbei werden die Leukozyten durch Erythrozyten aus dem axialen Blutstrom in Richtung des venösen Endothels verdrängt.41Daraufhin kommt es zum sogenannten Rollen. Verantwortlich dafür sind schwache Interaktionen zwischen den Leukozyten und den Endothelzellen. Diese Effekte sind gerade groß genug, um die Scherkräfte zu übertreffen und werden durch Selektine bzw. ihre Liganden vermittelt.
Das Rollphänomen von neutrophilen Granulozyten wurde anschaulich an künstlichen Oberflächen nachgestellt, welche mit P-Selektin42und E-Selektin43 beschichtet waren. Somit konnte deren Beteiligung an dieser Interaktion nachgewiesen werden.
Mikroskopische in vivo Untersuchungen an mesenterialen Gefäßen zeigten, dass monoklonale Antikörper gegen P-, E- und L-Selektin das Rollen der Leukozyten signifikant hemmen konnten.44,45,46Entsprechende Ergebnisse wurden später auch bei Untersuchungen an gendefizienten Mausmodellen beobachtet.47
In einem nächsten Schritt der Leukozytenadhäsion muss die Bindung derart verstärkt werden, dass die rollenden Leukozyten auf dem Endothel zum Stehen kommen. Hierbei spielen die Integrine eine entscheidende Rolle.
Wie schon erwähnt, werden Integrine auch basal exprimiert. Jedoch sind diese im unstimulierten Zustand der Zelle nicht aktiv bzw. affin. Sobald die Leukozyten durch geringe Konzentrationen von Zytokinen, chemoattraktiven Substanzen oder ähnlichem angeregt werden, wird die Oberflächendichte von L-Selektin drastisch verringert und sowohl die biologische Aktivität als auch die Oberflächendichte der Integrine erhöht.48 Diese aktivierenden Mediatoren werden durch Endothelzellen und andere Immunzellen ausgeschüttet. Der immunologische Aktivierungszustand des Neutrophilen folgt somit in gewisser Weise dem
Einleitung
Konzentrationsgradienten der ihn umgebenden Mediatoren. Vereinfacht kann man sagen, je mehr sich der Granulozyt dem inflammatorischen Focus annähert, desto stärker wird er aktiviert und ändert dementsprechend sein Oberflächenantigen-Expressionsmuster.
Neutrophile Granulozyten können darüber hinaus auch durch Interaktionen mit PECAM-1 aktiviert werden, was ebenfalls zu entsprechenden Veränderungen der Integrine führt.49Einmal aktiviert, führen die Integrine zur stationären Adhäsion der Leukozyten an den Endothelzellen. Die Endothelien weisen in ihrer Plasmamembran wiederum ein Oberflächenantigen-Expressionsmuster auf, das ebenfalls ihrem Erregungszustand angepasst ist. Daher verfügt eine stimulierte Gefäßwandzelle über eine sehr viel höhere Dichte von Adhäsionsmolekülen der Immunglobulin-Superfamilie als ihr unstimuliertes Pendant in einem Gebiet, das nicht unmittelbar mit dem entzündlichen Focus assoziiert ist. Folglich wird der Leukozyt zielgerichtet zum Entzündungsherd geführt und kann dann eine feste Bindung eingehen, um gegebenenfalls im weiteren Verlauf in das perivasale Gewebe überzutreten.
Diese starke Bindung ist an allen Leukozytenarten durch folgende Partner vermittelt: CD11a/CD18-ICAM1 sowie CD11a/CD18-ICAM2. Neutrophile Granulozyten interagieren zusätzlich über CD11b/CD18-ICAM1 und CD11c/CD18-ICAM1 mit den Endothelzellen. (vgl. Tabelle 1)
Dies wurde durch zahlreiche Untersuchungen an isolierten humanen Leukozyten und Endothelzellschichten bzw. zirkulierenden Leukozyten in postkapillaren Venolen evaluiert.21,39Hierbei wurde eine Vielzahl von Substanzen identifiziert, die regulatorisch in diesen, als feste Adhäsion bezeichneten, Schritt eingreifen. (s.
Tabelle 2)
Einleitung
Wirkort
Leukozyt Endothel
Proadhäsiv
Zytokine TNF- -1, -3, -5, -8 TNF- - , IL-1, IL4, IFN-
Lipide LPS, Leukotrien B4 LPS, Leukotrien B4
Peptide C3b, C5a, Neuropeptide Histamin Antiadhäsiv
Zytokine IL-4, -10, -13 IL-4, -10, -13 Prostaglandine Prostaglandin I2
Andere Adenosin, Stickstoffoxid Adenosin, Stickstoffoxid Tabelle 2 Übersicht der adhäsionsregulierenden Signalmoleküle (Auszug) 19
Im letzten Schritt der Leukozytenrekrutierung gelangen die aktivierten Leukozyten an den inflammatorischen Herd, indem sie durch die Endothelzellschicht migrieren. Dabei müssen die Leukozyten durch die Interzellular-Lücken des Endothels gelangen. Dies geschieht indem neue, starke Bindungen in progressiver Richtung geschaffen und in der Gegenrichtung gelöst werden. Hierbei dürfen die Bindungskräfte aber nicht zu stark sein, da hieraus sonst eine Immobilisation resultieren würde. In diesem Kontext zeigten Untersuchungen, dass die biologische Aktivität der Integrine auf den Leukozytenoberflächen nach der Stimulation durch PECAM-1 bzw. Zytokine sehr schnell wieder rapide abnimmt. Somit existiert eine Steuerungsgröße für die Schaffung und Lösung dieser starken Adhäsionskräfte.50 Des Weiteren spielt wahrscheinlich auch die herunterregulierte L-Selektin-Expression eine Rolle bei der Diapedese (Durchwanderung) der Leukozyten.5
Da die transendotheliale Migration durch monoklonale Antikörper gegen ICAM- 1 und CD11/CD18 effektiv gehemmt werden konnte, scheint dies ein weiterer Beweis für die Beteiligung dieser festen Adhäsionsmoleküle zu sein.51 Zusätzlich führt die einsetzende Kontraktion der Endothelzellen zu einer
Einleitung
Vergrößerung des interzellulären Spaltes und erleichtert somit die Diapedese der Leukozyten.52
Weitere elektronenmikroskopische Untersuchungen wiesen auch auf die Möglichkeit einer transendothelialen Diapedese auf transzellulärem statt interzellulärem Wege hin.53Die weitere Migration im subendothelialen Gewebe
1-Integrinen an Proteine der extrazellulären Matrix vermittelt.54
1.3 Sauerstoffradikalbildung
Nachdem der Leukozyt das Gefäßsystem verlassen hat und in den inflammatorischen Focus eingewandert ist, treten nun die direkt gegen den auslösenden Faktor gerichteten Abwehrmechanismen in den Vordergrund der Abwehrstrategie. Nebst der Phagozytose stellt die Bildung reaktiver Sauerstoffspezies (sog. oxidativer Burst) eine wichtige und entscheidende immunologische Funktion des neutrophilen Granulozyten dar.
Zur Bildung dieser Sauerstoffradikale wird die NADPH-Oxidase benötigt. Sie besteht aus einer membrangebundenen und vier zytosolischen Komponenten, welche nach Aktivierung des Neutrophilen an die Membraneinheit binden.55 Hierbei wird molekularer Sauerstoff mittels NADPH als Elektronendonor reduziert und die Sauerstoffradikale dann in intrazelluläre Kompartimente (Phagosomen) oder extrazellulär entlassen.56Diese Superoxidanionen können dann wiederum mit Wasser zu Wasserstoffperoxid reagieren. Im weiteren Verlauf reagiert das gebildete Wasserstoffperoxid intrazellulär mittels der Myeloperoxidase aus den azurophilen Vakuolen der Granulozyten mit freien Chloridanionen zu hypochloriger Säure, dem vermutlich eigentlichen zelltoxischen Effektor.57
Dabei wird jedoch auch deutlich, dass die reaktiven Sauerstoffspezies aufgrund fehlender Selektivität nicht nur antimikrobiell wirken, sondern auch umliegende Gewebe schädigen können und es auch tun. Konkret bedeutet dies, dass
Einleitung
neutrophile Granulozyten vor und bis zu sechs Stunden nach experimentell herbeigeführter Sepsis einen protektiven Effekt auf den Organismus haben.
Danach kommt es allerdings zu einer Akkumulation im geschädigten Gewebe, welche bis zu Organsdysfunktionen (MODS) und damit verbundener erhöhter Letalität führen kann.58Die Tatsache, dass dieser dramatische Verlauf eher selten auftritt, ist das Ergebnis eines komplexen immunregulatorischen Systems. Hierbei werden protektive Mechanismen der Immunabwehr gefördert und Kollateralschäden frühzeitig vermieden. Eine Säule dieser Regulatoren könnte der cholinerge antiinflammatorische Signalweg darstellen, welcher wiederum in Abschnitt 1.5 näher erläutert wird.
Ergänzend und rein informativ sei erwähnt, dass eine insuffiziente NADPH- Oxidase indes zu dem Krankheitsbild der chronischen Granulomatose führt.59
1.4 Die Proteinkinase C
Die Proteinkinase C (PKC) bezeichnet eine Enzymfamilie mit mehreren Subtypen. Dabei handelt es sich um wichtige intrazelluläre Signalmoleküle, die eine Rolle bei der Differenzierung, Proliferation und Aktivierung spezifischer Funktionen spielen. Diese Seronin/Threonin-Kinasen können sowohl von G- Protein-gekoppelten als auch von Tyrosinkinase- und wachstumshormonell- sensitiven Rezeptoren reguliert werden.60Derzeit sind zwölf Isoformen bekannt, welche wiederum in drei Hauptgruppen eingeteilt werden. Die Einteilung erfolgt auf Basis struktureller Gemeinsamkeiten. Hierbei ist die Präsenz der regulatorisch-wirksamen C1 bzw. C2Domäne entscheidend.
Zu der ersten Gruppe, den konventionellen Proteinkinase C Subtypen (cPKC), zählen die Proteinkinase Calpha, beta1, beta2 und -gamma. Sie besitzen sowohl die C1 als auch die C2Domäne, daher benötigen sie zur physiologischen Aktivierung Diacylglycerol (C1Domäne) und Calcium (C2
Domäne).
Einleitung
Eine zweite Gruppe bilden die neuen oder auch als nicht-konventionell bezeichneten Proteinkinasen C (nPKC), deren Vertreter sich aus Proteinkinase C
delta, epsilon, eta und theta zusammensetzen. Bei diesen Subtypen ist ebenfalls eine C1-Domäne vorhanden, jedoch fehlt die C2-Domäne. Somit fungiert diese Isoform Calcium-unabhängig.
Die dritte Gruppe bilden die atypischen PKC (aPKC). Ihr werden die Proteinkinase C iota, zeta und lambda zugeordnet. Hier finden sich strukturell weder eine C1 noch eine C2Domäne. In der Literatur wird zudem oft eine PKCmy [ ] beschrieben, die auch als Proteinkinase D1 geführt und den aPKC zugerechnet wird. 61,62
In der Vergangenheit konnten PKCalpha, beta1, beta2, delta und auch zeta in neutrophilen Granulozyten nachgewiesen werden.63 Die PKCIsoforme in neutrophilen Granulozyten sind unter anderem Teil des Signalwegs, der letztlich in der Produktion von Sauerstoffradikalen mündet. Hierzu phosphorylieren sie nach Aktivierung die zytosolische Untereinheit der NADPH-Oxidase (p47phox), was wiederum zur Aktivierung der NADPH-Oxidase führt.64Proteinkinase C
alpha wurde im Zusammenhang mit der gesteigerten Superoxidanion-Produktion nach PMA- und fMLP-Stimulus beschrieben.65Ebenfalls in die PMA-stimulierte Sauerstoffradikalbildung scheint die PKCdelta involviert zu sein. Zudem wurde ihrerseits eine erhöhte Aktivität nach TNF-alpha-Stimulus nachgewiesen.66,67 PMA ist ein PKC-Aktivator, dessen Funktionsweise sich durch die agonistische Bindung an die C1-Domäne der Kinase erklärt.68 Bei fMLP handelt es sich hingegen um ein bakterielles Peptid, welches von Bakterien als Initiatorsequenz der prokaryotischen Proteinsynthese gebildet wird.69 Dieses Peptid wirkt chemoattraktiv auf neutrophile Granulozyten, indem es an die beiden Rezeptoren
formyl peptide receptor (FPR) und formyl peptide receptor like 1 (FPRL1) bindet.70 Dadurch wird unter anderem die PKCdelta phosphoryliert, was wiederum über einige Zwischenschritte zur Aktivierung der NADPH-Oxidase führt.71
Einleitung
1.5 Das non-neuronale cholinerge System
Wie eingangs beschrieben, ist das Immunsystem ein essenzieller Faktor des Überlebens. Mindestens genauso wichtig ist jedoch auch die Kontrolle dieses Abwehrsystems, um Schäden am umliegenden Gewebe zu minimieren.
1975 schrieb Lewis Thomas in seinem Buch The lives of a cell:
When we sense lipopolysaccharide, we are likely to turn on every defense at our disposal; we will bomb, defoliate, blockade, seal off, and destroy all the tissues in the area. All of this seems unnecessary, panicdriven The self-disintegration of the whole animal that follows a systemic injection can be interpreted as a well- intentioned but lethal error. The mechanism is itself quite a good one, when used with precision and restraint. 72
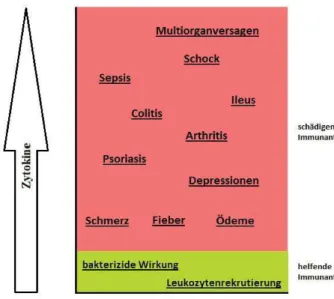
Abbildung 1 Folgen der unkontrollierten Zytokinausschüttung (nach Anlehnung an Tracey et al. 74)
Einleitung
Viele Symptome wie Fieber, Gewichtsverlust, Hypotonie, Koagulationsstörungen und andere Zeichen teils letaler Krankheitsbilder werden nicht direkt durch die Pathogene, sondern durch die humorale Immunantwort und inflammatorische Mediatoren wie IL-1, IL- ausgelöst.73(s. Abb. 1)
Der Tumornekrosefaktor-alpha ist ein Zytokin, das als Reaktion auf eine Infektion mit gram-negativen Bakterien bzw. deren Lipopolysaccharide (vorwiegend von Makrophagen) ausgeschüttet wird.74 Gleichzeitig ist dieser wichtige Bestandteil des Immunsystems von entscheidender Bedeutung für die Entstehung des septischen Schocks.75Gestützt werden diese Beobachtungen von Untersuchun
Mengen zu denselben hämodynamischen, metabolischen und immunologischen Konsequenzen führte, wie sie in einer Infektsituation zu beobachten sind.76Des Weiteren konnte diese Entwicklung durch pharmakologische TNF-Rezeptor- Blockade und bei gendefizienten Mäusen unterbunden werden.77,78
Wie schon im Abschnitt der Leukozytenadhäsion erwähnt, gibt es eine ganze Reihe humoraler antiinflammatorischer Mediatoren, die in der Regel die Wirkung proinflammatorischer Mediatoren auf einen sicheren und vorrangig schützenden Bereich beschränken.79 Hinzu kommen noch weitere eindämmende Mechanismen wie die hypothalamisch-hypophysäre-adrenale Achse.
Letztgenannte kann über eine kortikoide Immunsuppression den Abwehrmechanismen des Körpers entgegenwirken, sofern selbige eine schädigende Dynamik entwickeln. All diese Systeme reagieren jedoch langsam und entfalten eine systemische Wirkung. Sehr viel schneller und vor allem lokal, direkt am Ort der Entzündung, wirkt der sogenannte cholinergic antiinflammatory pathway (CAP).80
Das cholinerge System des Menschen wurde bis Ende des letzten Jahrtausends auf die neuronale Ebene beschränkt. Acetylcholin ist ein wichtiger Neurotransmitter. Über muskarinerge bzw. nikotinerge Acetylcholinrezeptoren wirkt es im menschlichen Nervensystem an Ganglien, Interneuronen und der
Einleitung
motorischen Endplatte. Von besonderer Bedeutung ist der zehnte Hirnnerv, der N. vagus, da er vom Kopf bis hin zur linken Colonflexur mäandert und dabei zahlreiche Gewebe hauptsächlich parasympathisch innerviert.
Studien haben gezeigt, dass die elektrische Stimulation des efferenten Vagusanteils zu einer verminderten, systemischen TNF-Ausschüttung während einer Endotoxämie (via LPS-Gabe) führt. In weiteren Studien wurde eine verminderte Zytokinaktivität sowie ein verbessertes Outcome nach physikalischer bzw. pharmakologischer Vagusstimulation bei experimentellen Sepsismodellen, hämorrhagischem Schock, Pankreatitis, Ileus, myokardialer Ischämie und Arthritis beobachtet.81,82,83,84,85,86,87
Die morphologischen Grundlagen dieser Beobachtungen stellt das non-neuronale cholinerge System dar. Zahlreiche Zellarten exprimieren abgekoppelt von neuronalen Einflüssen sowohl muskarinerge (mAChR) als auch nikotinerge (nAChR) Acetylcholinrezeptoren. Einen kurzen Überblick liefert die Tabelle 3.88
Zellart Acetylcholinrezeptor Literaturverw
eis Muskarinerg nikotinerg
humane
Endothelzellen M1 M5 3 5 7 2 4 89,90,91,92,93 Immunzellen
Mononukleäre
Zellen M1 M5 (variabel)
2, 4 5 6 7,
9 10, 2 4
(variabel, dominante
2, 5,
7)
94,95,96,97,98,
99,100,101,130 Eosinophile
Granulozyten M2, M3 3, 4 7
Neutrophile
Granulozyten M3 3 4 7 2 4
Einleitung
Tabelle 3 Übersicht Acetylcholinrezeptoren: Expressionsort und Subtypen88
Im letzten Jahrzehnt wurde der nikotinerge Acetylcholinrezeptor vom alpha-7- Bis heute konnte er auf beinahe allen Immunzelltypen nachgewiesen werden (s. Tabelle 3) und scheint eine regulatorische Funktion in der Immunantwort einzunehmen. Zunächst wurde
humane Makrophagen mittels Acetylcholin verringert werden konnte. Dieser Mechanismus schien post- -Bungarotoxin-sensitiven Nikotinrezeptor ausgelöst zu werden.101 Wang et al. gelang es 2001 diesen
98Zudem zeigte die Vagusstimulation bei endotoxämischen Mausmodellen eine verringerte proinflammatorische
-defiziente Knockout-Mäusen dieser Effekt nicht mehr erzielt werden konnte.98
ChR-Agonisten zum Einsatz. Nach Applikation konnten im Vergleich zu unbehandelten Gruppen signifikant - 1, IL-6 und HMGB-1 (high mobility group box 1) nachgewiesen werden.82,102 Eine grafische Zusammenfassung des cholinergen antiinflammatorischen Signalweges zeigt die Abbildung 2.
Makrophagen M2, M3 1 7 10
Mastzellen M1 M5 3 5 10
Einleitung
1.6 Fragestellung
Wie zuvor beschrieben, wurden schon zahlreiche Untersuchungen zur Aufklärung der Funktionsweise des CAP durchgeführt. Der nervus Vagus wurde sowohl physikalisch als auch pharmakologisch stimuliert. Die systemische Gabe von Cholinergika und direkten -Agonisten fand schon im Tiermodell Anwendung. Was jedoch unklar bleibt, ist die Wirkungsweise des CAP auf zellulärer Ebene abgekoppelt von neuronalen Einflüssen. Zwar wurden schon zahlreiche Rezeptortypen auf der Oberfläche von Immunzellen identifiziert, eine weitere Investigation des Zusammenspiels der einzelnen Komponenten blieb jedoch bisher aus.
Neutrophile Granulozyten sind ein wichtiger Bestandteil der zellulären Immunabwehr und übernehmen hierbei eine zentrale Rolle in der Abtötung und Beseitigung pathologischer Erreger. Aufgrund der zuvor beschriebenen Abbildung 2 Übersicht des CAP (in Anlehnung an Tracey et al.74)
Einleitung
pathophysiologischen Überlegungen und Hintergründe, versuchen wir die folgenden Fragen zu klären:
Wirken Acetylcholin, Nikotin oder Physostigmin in einer in vitro Studie modulatorisch auf die Sauerstoffradikalbildung von stimulierten und unstimulierten neutrophilen Granulozyten?
Ändert sich durch die Zugabe der genannten Cholinergika die Expression von CD11b oder CD62l auf neutrophilen Granulozyten?
Inwieweit haben die unterschiedlichen Aktivierungsmechanismen der getesten Stimulantien PMA, fMLP und TNF-alpha Einfluss auf eine etwaige cholinerge Immunmodulation an neutrophilen Granulozyten?
In einem weiteren Schritt wird dann der Effekt von Acetylcholin auf das Bindungsverhalten von neutrophilen Granulozyten an humanen, umbilical- venösen Endothelzellen (HUVEC) mit Fokus auf die endotheliale Antigenexpression näher beleuchtet. Diese Endothel-Granulozyten-Interaktion gilt als Grundvoraussetzung der granulozytären Immunantwort. Hierbei eignen sich die endothelialen Adhäsionsmoleküle CD54 und CD106 als Messparameter des immunologischen Funktionszustandes der Endothelzellen, da diese mit den zuvor genannten Oberflächenantigenen der neutrophilen Granulozyten interagieren und somit den Übertritt der immunologischen Effektorzellen in das perivasale Gewebe ermöglichen. Dadurch beabsichtigen wir die folgenden Fragen zu klären:
Wirkt Acetylcholin modulatorisch auf das CD54- bzw. CD106- Expressionsverhalten von IL1-stimulierten oder unstimulierten humanen, umbilical-venösen Endothelzellen?
Wirken sich etwaige Beobachtungen, bzgl. eines cholinergen Effektes auf die Antigenexpression der neutrophilen Granulozyten oder der humanen,
Einleitung
umbilical-venösen Endothelzellen, tatsächlich auf die Leukozyten- Endothel-Interaktion aus?
Hierbei finden alle Untersuchungen in vitro statt, da der Einfluss neuronaler Netzwerke ausgeschaltet und isoliert die zelluläre Ebene des CAP untersucht werden soll.
Material und Methoden
2 Material und Methoden 2.1 Material
2.1.1 Geräte
Name Hersteller
Heraeus Megafuge 1.0 R Heraeus Instruments GmbH, Osterode, Deutschland
Heraeus Biofuge pico Heraeus Instruments GmbH, Osterode, Deutschland
HAAKE Schüttelwasserbad SWB25
Thermo Fischer Scientific Inc., Waltham, Massachusetts, USA
LabTherm Schüttelgerät Adolf Kühner AG, Basel, Schweiz BD FACSCalibur
Durchflusszytometer
BD Biosciences , Franklin Lakes, New Jersey, USA
MS 2 Minishaker IKA Werke GmbH & Co. KG, Staufen, Deutschland
Varioskan Flash Thermo Fischer Scientific Inc., Waltham, Massachusetts, USA
LaminAir HB2448 Heraeus Instruments GmbH, Osterode, Deutschland
accuJet pro Brand GmbH & Co KG, Wertheim, Deutschland
HandyStep Pipette Brand GmbH & Co KG, Wertheim, Deutschland
Eppendorf Research 20 l, 200 l, 1000 l
Eppendorf AG, Hamburg, Deutschland
FunctionLine CO2-Inkubator Heraeus Instruments GmbH, Osterode, Deutschland
Glaspipette 5ml, 10ml Sarstedt AG, Nümbrecht, Deutschland Tabelle 4 verwendete Geräte
Material und Methoden 2.1.2 Verbrauchsmaterial
Tabelle 5 verwendetes Verbrauchsmaterial
2.1.3 Reagenzien
Name Hersteller
Dulbeccos PBS w/o Ca+Mg Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
Carboxy SNARF®-1 Acetoxymethyl Ester Acetate (Seminaphtharhodafluor)
Invitrogen Corporation, Grand Island, New York, USA
Dihydrorhodamine 123 Cayman Chemical Company, Michigan , USA
Name Hersteller
Pipettenspitzen, unsteril, 10 l, 200 l, 1000 l
Sarstedt AG, Nümbrecht, Deutschland
FACS-Röhrchen, 5ml Sarstedt AG, Nümbrecht, Deutschland Reagenzröhrchen, 10ml Sarstedt AG, Nümbrecht, Deutschland SafeSeal-Reagiergefäß, 1,5ml,
2ml
Sarstedt AG, Nümbrecht, Deutschland
Parafilm M Laboratory Film Pechiney Plastic Packing, Chicago, USA 96-well Mikrotestplatte Sarstedt AG, Nümbrecht, Deutschland Peha-Soft, Latex-Handschuhe Paul Hartmann AG, Heidenheim,
Deutschland Combitips plus® biopur, 0,5ml,
10ml, 50ml
Eppendorf AG, Hamburg, Deutschland
Multiwell 24-well Platte BD Biosciences, Franklin Lakes, New Jersey, USA
Material und Methoden
Acetylcholinchlorid, 98+% Alfa Aesar GmbH & Co KG, Karlsruhe, Deutschland
humaner TNF- PeproTech GmbH, Hamburg, Deutschland Phorbol-12-myristate-13-acetate
(PMA)
Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
N-Formyl-L-methionyl-L-leucyl-L- phenylalanin (fMLP)
Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
Propidiumiodid SERVA Electrophoresis GmbH, Heidelberg, Deutschland
Physostigminsalicylat Dr. Franz Köhler Chemie GmbH, Bensheim, Deutschland
FITC anti-human CD11b BioLegend, San Diego, California ,USA FITC anti-human CD62L Beckman Coulter GmbH, Krefeld,
Deutschland
Histopaque-1077 Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
Dextran T500 Pharmacia LKB, Uppsala, Schweden NaCl 0,9% B. Braun B. Braun Melsungen AG, Melsungen,
Deutschland
PhosStop (Phosphatase-Inhibitor) Roche Diagnostics GmbH, Mannheim, Deutschland
cOmplete,Mini,EDTA- free,EASYpack (Protease-Inhibitor)
Roche Diagnostics GmbH, Mannheim, Deutschland
Triton X-100 Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
Ethylendiamintetraacetat (EDTA) Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
Material und Methoden
Desoxycholsäure Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
Natriumdodecyl-sulfat Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
Ammoniumchlorid Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
Natriumhydrogencarbonat Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
Aqua dest. Laboreigene Herstellung
Pestanal®, (-)-Nikotin Fluka Chemie AG, Buchs, Schweiz
BD FACSFlow BD Biosciences, Franklin Lakes, New Jersey, USA
BD FACSRinse Solution BD Biosciences, Franklin Lakes, New Jersey, USA
Endothelial cell growth medium Provitro GmbH, Berlin, Deutschland Supplement kit for endothelial cell
groth medium, FCS-Kit
Provitro GmbH, Berlin, Deutschland
0,5%Trypsin/EDTA (10x) Life Technologies Ltd., Paisley, UK Hanks BSS w/o
Calcium/Magnesium, w/o phenol red
PAA Laboratories GmbH, Pasching, Österreich
Human Interleukin-1beta EnzoLifesciences
FITC anti-human CD54 BioLegend, San Diego, California, USA PE anti-human CD106 BioLegend, San Diego, California, USA Gentamicin 10mg/ml PAA Laboratories GmbH, Pasching,
Österreich
Gelatine from bovine skin, Type B Sigma-Aldrich Chemie GmbH, Steinheim, Deutschland
Material und Methoden
PKC kinase activity kit Enzo Life Sciences GmbH, Lörrach, Deutschland
Humane PKC-delta recombinant Enzo Life Sciences GmbH, Lörrach, Deutschland
Tabelle 6 verwendete Reagenzien
2.1.4 Puffer-Lösungen RIPA-Puffer:
Lösungsmittel: Dulbeccos PBS (phosphat buffered saline) 0,1% Natriumdodecylsulfat
0,5% Natrium-Desoxycholsäure 1% Triton X-100
Protease-Inhibitor (Dosierung nach Herstellerangaben) Phosphatase-Inhibitor (Dosierung nach Herstellerangaben) RBCL-Puffer:
Lösungsmittel: destilliertes Wasser 0,9% Ammoniumchlorid
0,2% Natriumhydrogencarbonat 30% EDTA (Ethylendiamintetraacetat) 2.2 Methoden
2.2.1 Probanden
Die neutrophilen Granulozyten wurden gemäß unten stehendem Protokoll aus dem Vollblut von freiwilligen, aufgeklärten Probanden isoliert. Alle Probanden waren männlich, Nichtraucher, Nichtdiabetiker und zum Zeitpunkt der Blutentnahme augenscheinlich gesund. Des Weiteren war die Krankheitsanamnese der letzten 14 Tage negativ. Gleiches galt für die Einnahme von Medikamenten, die zur Gruppe der nichtsteroidalen Antirheumatika und den
Material und Methoden
Antihistaminika gehören. Je Versuchsanordnung wurden zehn Probanden akquiriert, deren Alter zwischen 20 und 26 Jahren lag. Etwaige Abweichungen hiervon werden an den entsprechenden Stellen ausgewiesen.
2.2.2 Getestete Substanzen und Konzentrationen
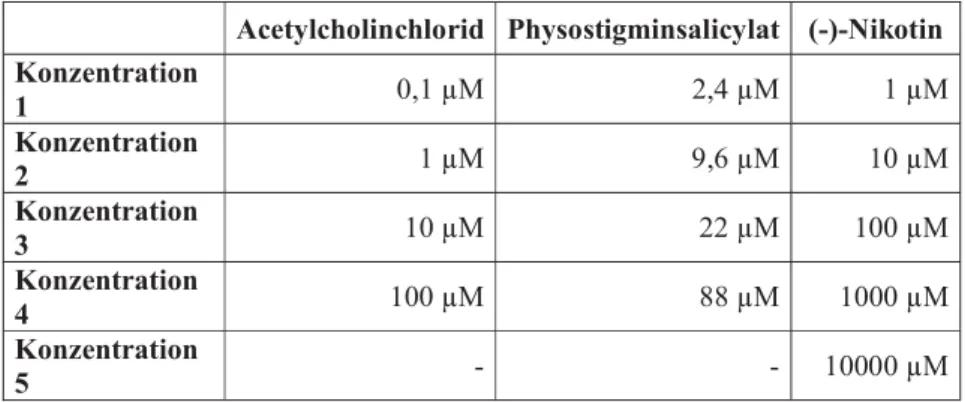
In dieser Studie wurde die Wirkung von Acetylcholinchlorid, (-)-Nikotin und Physostigminsalicylat untersucht. Bei den Messungen zur Sauerstoffradikalbildung, der CD11b- und der CD62l-Expression kamen folgende finale Konzentrationen zum Einsatz:
Acetylcholinchlorid Physostigminsalicylat (-)-Nikotin Konzentration
1 0,1 M 2,4 M 1 M
Konzentration
2 1 M 9,6 M 10 M
Konzentration
3 10 M 22 M 100 M
Konzentration
4 100 M 88 M 1000 M
Konzentration
5 - - 10000 M
Tabelle 7 Übersicht der getesteten Substanzen und Konzentrationen102103 Während der Versuchsreihen zur CD54- und CD106-Expression an humanen, umbilical-venösen Endothelzellen wurde ausschließlich Acetylcholinchlorid in den Konzentrationen 0,1 M, 1 M, 10 M, 100 M und 1000 M genutzt. Da die Versuche mit dem ELISA zur Quantifizierung der Proteinkinase C Aktivität auf den Ergebnissen der vorangehenden Durchflusszytometrie beruhten, fand hier wiederum nur Physostigminsalicylat in den Konzentrationen 2,4 M, 9,6 M, 22
M, 88 M und zusätzlich 242 M Anwendung.
Die untersuchten Physostigminkonzentrationen entstammten Versuchen mit neutrophilen Granulozyten am Rattenmodell107, während die eingesetzten Nikotinmengen auf Publikationen von Pabst et al.104sowie Speer105basieren. Bei
Material und Methoden
den Acetylcholin-Versuchen waren uns indes keine Publikationen mit ähnlichem Versuchsaufbau bekannt, daher wurden die eingesetzten Acetylcholin-Mengen frei gewählt und möglichst breit gefächert.
Sofern nicht anders beschrieben, wurden alle Versuche im Doppelansatz durchgeführt.
2.2.3 Isolation der neutrophilen Granulozyten 2.2.3.1 Durchflusszytometrie
Zur Gewinnung von humanem Vollblut wurde den Probanden etwa 15ml Blut aus den antekubitalen Venen entnommen und in drei 5,5ml Lithium-heparinisierten Blutentnahme-Röhrchen gesammelt. Danach wurden je 3ml eines Histopaque®- Gradienten (1077g/ml) mit 3ml des heparinisierten Vollblutes überschichtet und 90 Minuten bei Raumtemperatur inkubiert. Nachdem sich eine untere erythrozytenreiche, eine mittlere leukozyten- und lymphozytenreiche sowie eine obere Histopaque-Schicht voneinander getrennt hatten, wurde je 1ml der mittleren Schicht abpipettiert und in einem 5ml FACS-Röhrchen gesammelt. Der Rest wurde verworfen.
2.2.3.2 ELISA
Die Blutentnahme erfolgte analog zu den durchflusszytometrischen Versuchsaufbauten mit dem einzigen Unterschied, dass für die ELISA-Versuche 20ml Blut entnommen wurden.
Es wurden ebenfalls 3ml eines Histopaque®-Gradienten (1077g/ml) mit 3ml des heparinisierten Vollblutes überschichtet, dann allerdings für 45 Minuten bei 4°C und 400x g zentrifugiert. Die beiden oberen Schichten, bestehend aus Plasma und
Material und Methoden
Monozyten bzw. dem Histopaque®, wurden verworfen und der Rest (vornehmlich Granulozyten und Erythrozyten) in einer 6%igen Dextran-Lösung (aufgelöst in 0,15M NaCl-Lösung) resuspendiert sowie mit weiteren 2ml Dulbeccos PBS (Phosphatpuffer) homogenisiert.
Nach 20 Minuten Inkubation im 37°C Wasserbad wurde der Überstand gesammelt und für 10 Minuten bei 4°C und 270x g zentrifugiert. Der Überstand wurde verworfen und das Zellpellet in 2ml RBCL-Puffer (red blood cell lysis puffer) aufgelöst. Nach achtminütiger Inkubation bei Raumtemperatur und Dunkelheit wurde die Suspension wiederum mit 2ml PBS homogenisiert und für 5 Minuten bei 4°C und 350x g zentrifugiert.
Das Zellpellet wurde daraufhin mit 1ml PBS gewaschen, wiederum zentrifugiert (3 Minuten, 4°C, 350x g) und in 800 l PBS resuspendiert.
Das so gewonnene Granulozyten-Konzentrat wurde nun lysiert. Hierzu wurde es zunächst auf 2ml Eppendorf-Cups verteilt und bei 16200x g für 3 Minuten zentrifugiert. Der Überstand wurde verworfen und das Zellpellet mit 100 l RIPA- Puffer überschichtet. Nach sorgfältiger Durchmischung mittels eines Vortex, wurde die Lösung einem dreimaligen Einfrier-Auftauzyklus in flüssigem Stickstoff unterzogen.
Zum Schluss wurden die nun lysierten Zellen bei 16200x g und 4°C für 30 Minuten zentrifugiert, die flüssige Phase mitsamt den darin enthaltenen Proteinen abgehoben und bis zur weiteren Verarbeitung bei -86°C eingelagert.
2.2.4 Messung der Sauerstoffradikalbildung 2.2.4.1 Theoretische Grundlagen der Messung
Wie schon beschrieben, gehört die Sauerstoffradikalbildung zu den wichtigsten Abwehrmaßnahmen der neutrophilen Granulozyten. Die durch die NADPH-
Material und Methoden
Oxidase produzierten Sauerstoffradikale verbinden sich mit intrazellulärem Wasser zu Wasserstoffperoxid (H2O2).
Als Messsubstanz wird das nichtfluoreszierende, zellmembranpermeable Dihydrorhodamin-123 (DHR) eingesetzt. Es wird durch das anfallende Wasserstoffperoxid zu Rhodamin-123 oxidiert einem bei 488nm Wellenlänge durch Licht anregbaren Fluoreszenzfarbstoff. Hierbei ist das gebildete Rhodamin- 123 annähernd proportional zu dem entstehenden Wasserstoffperoxid und somit auch zu den von der NADPH-Oxidase produzierten reaktiven Sauerstoffspezies.
Im Vergleich zu 2',7'-Dichlorofluorescindiacetat gilt die Messung der Sauerstoffradikalbildung mittels Dihydrorhodamin-123 als weitaus sensitiver.106
2.2.4.2 Ablauf der Messung
Zur Messung der Sauerstoffradikalbildung wurden FACS-Röhrchen mit 950 l Dulbeccos PBS befüllt und anschließend je 20 l leukozytenreiches Plasma hinzupipettiert. Danach wurden je 10 l Dihydrorhodamin 123 (DHR) sowie 10 l SNARF® hinzugefügt und für 10 Minuten in einem 37° C warmen Wasserbad inkubiert. Dies entspricht einer finalen DHR- und SNARF-Konzentration von je 100nmol/l.
In einem nächsten Schritt erfolgte die Zugabe von 10 l der zu testenden Substanz in der entsprechenden Konzentration (s. Abbildung 3) und daraufhin wiederum eine 10-minütige Inkubation im 37°C-Wasserbad. Nach 5 Minuten der Wasserbad-Inkubation wurden der fMLP plus -Gruppe 10 l humaner
Tumornekrosefaktor- -Konzentration
von 10ng/ml ergab.
Material und Methoden
Ohne Stimulus
PMA fMLP fMLP plus
Ohne Testsubstanz Konzentration 1 Konzentration 2 Konzentration 3 Konzentration 4 (evtl. Konzentration 5)
Abbildung 3 Schema des Probenansatzes und verwendete Stimulantien
Zuletzt wurden entsprechend Abbildung 3 die jeweiligen Stimuli hinzupipettiert.
Die entsprechenden finalen Konzentrationen beliefen sich für PMA und für die beiden Gruppen mit fMLP-Stimulus auf je 100 nM pro Ansatz.
Alle Proben wurden 15 Minuten im 37°C-Wasserbad inkubiert und danach die Reaktion unverzüglich durch Lagerung auf Eis abgestoppt. Nach Zugabe von 10 l Propidiumiodid (finale Konzentration: 150nM) wurden die Proben als Vorbereitung auf die durchflusszytometrischen Messungen bei 400x g und 4°C für 3 Minuten zentrifugiert, der Überstand abgekippt und das Zellpellet in 200 l Dulbeccos PBS resuspendiert. Im Anschluss erfolgten die FACS-Messungen.
Material und Methoden 2.2.5 Messung der CD11b- und CD62l-Expression
Die Messung der Oberflächenantigen-Expression erfolgte fast analog zu der oben beschriebenen Vorgehensweise. Es wurden FACS-Röhrchen mit 980 l Dulbeccos PBS befüllt und 20 l des leukozytenreichen Plasmas hinzugefügt.
Direkt hierauf erfolgte die Applikation der zu testenden Substanzen nach o.g.
Schema und eine 10-minütige Inkubationsphase. Darauf folgend wurden wiederum die Stimuli hinz
nur PMA sowie fMLP Anwendung fanden. Die Konzentrationen und ebenso die anschließende Inkubationszeit von 15 Minuten orientierten sich an dem Versuchsablauf zur Sauerstoffradikal-Messung.
Im Anschluss wurden alle Proben bei 400x g und 4°C für 3 Minuten zentrifugiert.
Der Überstand wurde verworfen, 10 l fluorescein-isothiocyanate (FITC)- konjugierte CD11b- bzw. CD62l-Antikörper (je nach Ansatz) zu allen Proben hinzupipettiert und das Zellpellet in Suspension gebracht. Anschließend fand eine 30-minütige Inkubationsphase bei 4°C statt.
Schlussendlich wurden die Proben mit 1ml Dulbeccos PBS verdünnt, bei 400x g für 3 Minuten zentrifugiert, der Überstand wiederum verworfen und das Zellpellet in 200 l PBS resuspendiert. Im Anschluss erfolgten auch hier die FACS- Messungen.
2.2.6 Durchflusszytometrie (FACS-Messung)
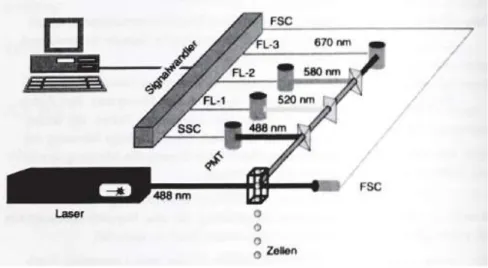
Die Durchflusszytometrie ist eine elektro-optische Messmethode bei der sowohl Lichtstreuung als auch Fluoreszenzen eine Rolle spielen.
Während der Messung werden die Zellproben mittels eines Hüllstroms verdünnt und fokussiert, so dass die Zellen kettenförmig, nacheinander einen Messpunkt passieren. An diesem Messpunkt trifft ein Laserstrahl rechtwinklig auf die Proben. Dies hat zur Folge, dass dieser in einer für viele Zellen charakteristischen
Material und Methoden
Art und Weise gestreut wird. Hierbei unterscheidet man die Vorwärtslichtstreuung (front scatter, FSC) und die Seitwärtsstreuung (side scatter, SSC), welche Auskunft über Zellgröße (FSC) bzw. Zellgranularität (SSC) geben. Zeitgleich erfolgt durch die Belichtung mit dem monochromatischen Licht des Laserstrahls eine Anregung der evtl. verwendeten Fluoreszenzfarbstoffe.
Durch die Verwendung unterschiedlicher Farbstoffe für unterschiedliche Fragestellungen
(s. Tabelle 8) ist es notwendig, die verschiedenen Emissionsspektren aufzutrennen.
Hierzu werden Bandpassfilter und Farbteilerspiegel eingesetzt. Sie leiten die optischen Signale zu Photomultipliern (Detektoren), welche wiederum spezifische Lichtspektren detektieren.
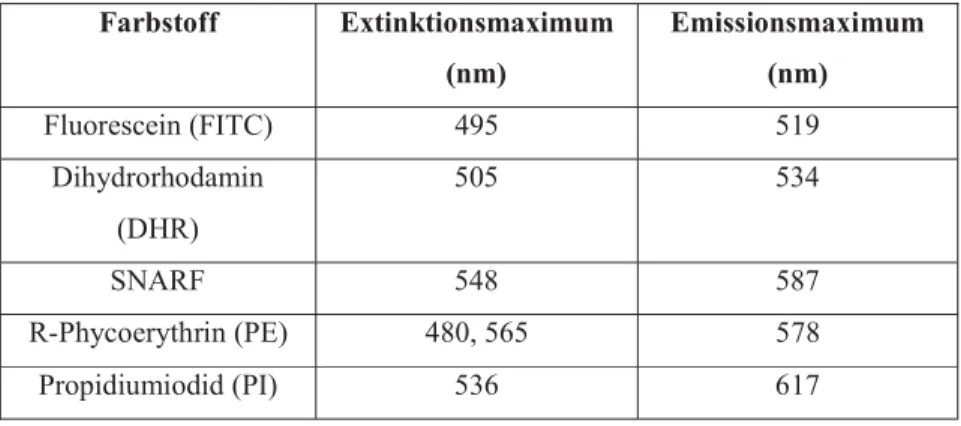
Farbstoff Extinktionsmaximum (nm)
Emissionsmaximum (nm)
Fluorescein (FITC) 495 519
Dihydrorhodamin (DHR)
505 534
SNARF 548 587
R-Phycoerythrin (PE) 480, 565 578
Propidiumiodid (PI) 536 617
Tabelle 8 Übersicht der Fluoreszenzfarbstoffe und ihrer Farbspektren123
Dadurch ist es beispielsweise möglich, zwei verschiedenartige Oberflächenproteine gleichzeitig auf einer Zelle sichtbar zu machen. In der Praxis verwendet man bei dieser Fragestellung für Antigen 1 einen Antikörper mit Fluoreszenzfarbstoff 1 und parallel für Antigen 2 einen Antikörper mit
Material und Methoden
Fluoreszenzfarbstoff 2. Hierbei können jedoch Probleme auftreten, wenn sich Emissionsspektren überschneiden. Diese kann man aber durch Kompensationsmechanismen weitgehend ausgleichen.
Der Nomenklatur folgend werden die einzelnen Detektoren nach zunehmendem Abstand zum anregenden Wellenlängenbereich geordnet und dementsprechend mit FL1, FL2 und so weiter benannt. FL steht hierbei lediglich für fluorescent light. In unserem Fall handelte es sich bei dem Laser um einen Argonlaser mit einem Wellenlängenbereich von 488nm (blau-grün). Der Detektor FL1 war für blaues Licht, FL2 für orangenes und FL3 für das rote Lichtspektrum zuständig.
An die Detektoren ist ein Signalwandler angeschlossen. Hier werden aus den optischen Signalen elektrische Impulse erzeugt und dann, mittels eines mit dem Zytometer verbundenen Computersystems, in multiparametrischen Dateien abgelegt. Dank der direkten Speicherung der reinen Messdaten kann ein Datensatz für unterschiedliche Fragestellungen herangezogen werden.
Abbildung 4 zeigt den schematischen Aufbau eines solchen Durchflusszytometers. Hierbei wird der Strahlengang und dessen Aufspaltung in die einzelnen Emissionsspektren deutlich.
Abbildung 4 entnommen aus Zelluläre Diagnostik von Ulrich Sack 123
Material und Methoden
2.2.7 Quantifizierung der Proteinkinase C – Aktivität 2.2.7.1 Der ELISA
Die Messung der Proteinkinase C Aktivität erfolgte mittels eines speziellen ELISA-Kits.
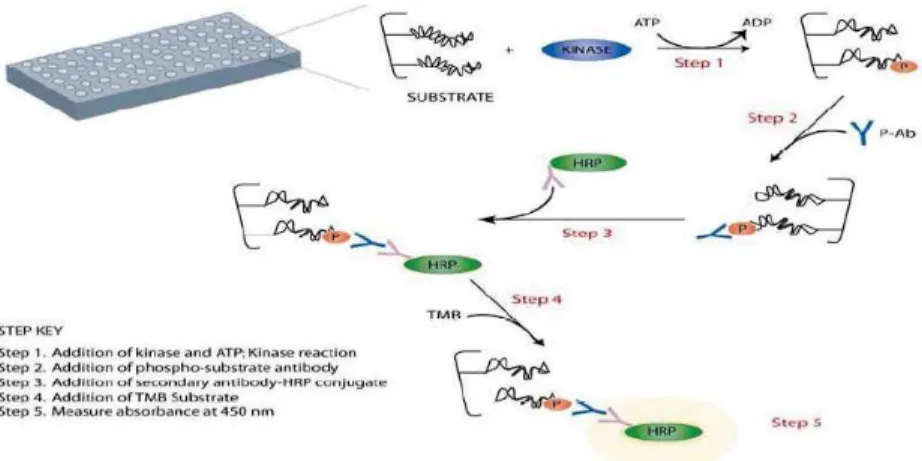
ELISA ist die Abkürzung für enzyme-linked immunosorbent assay. Hierbei handelt es sich um eine mehrstufige Antigen-Antikörper-Reaktion mit konjugierten Enzymen, deren Substratumsatz schlussendlich photometrisch gemessen und somit indirekt Rückschlüsse auf die ursprünglich vorhandene Substratmenge, Enzymaktivität oder ähnliches gezogen werden können. Im Folgenden sei die Funktionsweise des von uns genutzten PKC-Activity KIT erläutert.
Die dem ELISA beiliegende 96-Well Mikrotiterplatte wurde herstellerseitig mit einem phosphorylierbaren Peptid beschichtet. Nach Einpipettieren des zu testenden Materials (Proteinproben) in die Wells, wurde die Reaktion durch Zugabe einer definierten Menge ATP gestartet. Nach einer 90-minütigen Inkubationsphase bei 30°C und kontinuierlichem Schwenken mit 60 Umdrehungen pro Minute, wurde die Mikrotiterplatte gewaschen und dadurch die Reaktion abgestoppt.
Je nach Aktivitätsgrad und Menge des in der Proteinprobe enthaltenen PKC- Anteils, befand sich nun mehr oder weniger phosphoryliertes, immobilisiertes Substrat am Boden der einzelnen Wells.
Im nächsten Schritt wurde die Mikrotiterplatte mit einem Primärantikörper gegen das phosphorylierte Substrat (=Produkt) beimpft und für 60 Minuten bei Raumtemperatur unter ständigem Schwenken ruhen gelassen. Nach Ablauf der Zeit wurden die Wells entleert, sowie überschüssige Antikörpermengen durch mehrfaches Spülen entfernt.
Danach wurden die Wells mit einem Sekundärantikörper beschichtet, welcher spezifisch an den zuvor erwähnten Primärantikörper bindet und an dem zusätzlich
Material und Methoden
ein Enzym konjugiert ist. Bei diesem Enzym handelte es sich um die Meerrettichperoxidase (horseradish peroxidase, HRP).
Nach weiteren 30 Minuten Inkubation bei Raumtemperatur und gelegentlichem Schwenken wurden die Wells im Zuge eines mehrmaligen Waschvorganges wieder entleert und Überschüsse weggespült. In den Wells der Mikrotiterplatte befanden sich nun das phosphorylierte, fest an der Platte haftende Substrat, der daran gebundene Primärantikörper sowie der an den Primärantikörper gebundene Sekundärantikörper mitsamt der daran konjugierten HRP. (s. Abbildung 5)
Abbildung 5 Prinzip des ELISA (Auszug aus beiliegender
Bedienungsanleitung der Fa. Enzo Life Sciences GmbH)
Nun erfolgte die Zugabe von 3,3',5,5'-Tetramethylbenzidin (TMB) und Wasserstoffperoxid - den Substraten der HRP. Das farblose TMB dient hierbei als Elektronendonor für die Reduktion von Wasserstoffperoxid zu Wasser durch die Peroxidase und wurde dadurch selbst zu einem blau-leuchtenden Produkt oxidiert.
Material und Methoden
Die Reaktion wurde entsprechend der Anleitung nach 30 bis 60 Minuten (je nach Färbungsgrad) mit Schwefelsäure abgestoppt, wodurch sich eine Gelbfärbung ergab, die proportional zu der ursprünglichen PKC-Phosphortransferase-Aktivität war und bei 450nm Wellenlänge photometrisch gemessen werden konnte.
2.2.7.2 Bestimmung der Proteinkonzentration
Die Proteinkonzentrationsbestimmung erfolgte mittels der BCA-Methode.
Hierbei steht BCA für bicinchoninic acid (Bicinchoninsäure) und bezeichnet ein Verfahren, das auf der Biuretreaktion basiert. Dies bedeutet, dass jeweils zwei Peptidbindungen mit einem Cu² -Ion einen blauvioletten Komplex bilden, sofern eine alkalische Umgebung vorherrscht. Der Komplex kann dann bei 540nm photometrisch gemessen werden.
Um eine Quantifizierung zu erreichen, wurde neben den Proben auch eine Standardreihe mit bekannten Proteinkonzentrationen gemessen. Diese bestand aus neun Werten und erstreckte sich von null bis zwei Milligramm pro Milliliter.
Aus diesen Werten konnte dann eine Kurve erzeugt und die photometrisch gewonnenen Werte der Proben über selbige gelegt werden.
Die eingesetzten Proteinmengen ließen sich nun aus dem verwendeten Volumen (20 l) und den jeweiligen abgelesenen Proteinkonzentrationen errechnen.
2.2.7.3 Probandenproteine
Für den ersten PKC-Versuch wurde isoliertes Protein aus Probandenmaterial (n=8) genutzt. Das Isolationsverfahren ist ausführlich in 2.2.3.a beschrieben.
Der ELISA wurde gemäß den herstellerseitigen Angaben vorbereitet und anschließend pro Probandenprobe zehn Wells mit je 30 l Probenmaterial befüllt.
Zusätzlich wurden zwei Wells mit je 30 l der dem Kit beigefügten positiv
Material und Methoden
Kontrolle (PKC ells als Leerwert frei gelassen. Die Inkubation mit den entsprechenden Physostigminsalicylatkonzentrationen (2,4
weiligen Well, wobei zwei Wells je Proband unbehandelt blieben. Nach Ablauf der Inkubationszeit wurde die Reaktion durch ATP-Zugabe gestartet. Alle nachfolgenden Schritte entsprechen dem vorher beschriebenen Ablauf und wurden gemäß Handbuch mit den mitgelieferten Chemikalien durchgeführt.
Parallel zu dem PKC-ELISA fand eine Proteinkonzentrationsbestimmung (BCA- Messung) statt, um Kenntnis über die eingesetzte Probenmenge zu erhalten.
Hierzu wurden alle Probandenproben im Dreifachansatz gemessen.
2.2.7.4 Gereinigte Proteinkinase C - delta
Bei dem folgenden Versuch wurden statt der Probandenproben, welche ein Konvolut verschiedener Enzyme und sonstiger Zellbestandteile enthielten, reine Proteinkinase C des Deltasubtyps verwendet.
Eine BCA-Messung schien uns bei diesem Versuchsaufbau überflüssig, da die Proteinkonzentration der gekauften Proteinkinase vom Hersteller angegeben wurde. Die eingesetzten Proteinmengen betrugen 0,2 , 2 und 20 ng pro Well und wurden vor der ATP-Zugabe mit Physostigminsalicylatkonzentrationen von 2,4 - 88 M für 15 Minuten bei Raumtemperatur inkubiert.
Zusätzlich zu dem Leerwert und der Positivkontrolle (im Kit enthalten) wurde Staurosporin eingesetzt. Hierzu wurden pro Proteinmenge je zwei Wells mit 25 g dieses PKC-Inhibitors124 versetzt und ebenfalls für 15 Minuten bei Raumtemperatur inkubiert.
Alle Messungen fanden im Doppelansatz statt.
Material und Methoden
2.2.8 Versuche mit HUVEC 2.2.8.1 An- und Aufzucht
Die Endothelzellen aus den Umbilicalvenen menschlicher Nabelschnüre wurden uns im kryokonservierten Zustand von der Abteilung für Herz-Thorax-Chirurgie des Universitätsklinikums Regensburg zur Verfügung gestellt. Deren Extraktion fand mittels Kollagenase nach dem üblichen Protokoll statt107.
Zunächst wurden für die Anzucht T25-Kulturflaschen verwendet. Da Endothelzellen eine Fläche zum Anhaften benötigen, wurden die Flaschen mit 0,2%iger Gelatine aus Rinderhaut beschichtet, anschließend mindestens eine halbe Stunde bei 37°C inkubiert und danach der Überstand abgesaugt.
Um die Zellen überhaupt anzüchten zu können, wurden sie kurz im 37°C- Wasserbad angetaut, so dass sie sich aus dem Kryoröhrchen lösen ließen. Danach wurden die gefrorenen Zellen in 4°C warmes Kulturmedium überführt um vollständig aufzutauen. Darauf folgte eine Zentrifugation bei 400x g und 4°C für 5 Minuten, um das zellschädigende Lösungsmittel Dimethylsulfoxid (DMSO) zu entfernen. Der Überstand wurde steril abgesaugt und das Zellpellet in Endothel- Kulturmedium (mit beiliegendem Supplement-Kit versetzt) resuspendiert. Das
Supplement-Kit besteht aus mehreren Komponenten und ist jedem verwendeten Endothel-Kulturmedium beigefügt worden.
Konkret besteht das Supplement-Kit aus:
(Konzentrationen pro ml Medium) fetales Kälberserum 00.02 ml
ECGS/H (3 mg/ml) 04.00 l (endothelial cell growth supplement/Heparin)
humaner EGF 00.10 ng (endothelial growth factor) humaner bFGF 01.00 ng (basic fibroblast growth factor)
Material und Methoden
Hydrocortison
Gentamicin
Amphotericin B 50.00 ng L-Glutamin (2mM) 00.02 ml
Nun konnte die Zellsuspension in der vorbereiteten T25-Flasche ausgesät und bei 37°C inkubiert werden. Alle zwei bis drei Tage fand ein Wechsel des Kulturmediums statt.
Sobald circa 80% Konfluenz erreicht waren, wurden die Zellen gesplittet und in zwei ebenfalls gelatinierte T75-Zellkulturflaschen überführt. Hierzu wurde 0,5%iges Trypsin (+0,2% EDTA) in einem 1:10 Verhältnis mit Dulbeccos PBS verdünnt und in die T25-Kulturflaschen gegeben. Sofern sich im Lichtmikroskop eine vollständige Ablösung der Zellen vom Untergrund nachweisen ließ, wurde die gesamte Zellsuspension der Kulturflasche entnommen und sofort für 5 Minuten bei 400x g und 20°C zentrifugiert. Danach wurde der Überstand verworfen, das Pellet in 10ml Kulturmedium resuspendiert und je 5ml zur erneuten Aussaat in die vorbereiteten T75-Kulturflaschen verwand, welche dann wiederum im 37°C Inkubator aufbewahrt wurden. Auch hier fand wiederum alle zwei bis drei Tage ein Mediumwechsel statt.
Ab einem einheitlichen Zellrasen von circa 80% Konfluenz wurden die HUVECs wiederum nach obigen Schema aus der T75-Flasche gelöst und nach der Zentrifugation sowie der Resuspension in 10ml Nährmedium in einem 50ml
Falcon belassen. in eine sog. Neubauer-
Zählkammer überführt, um Aufschluss über die tatsächliche Zellkonzentration zu erhalten. Hierzu verfügt diese Zählkammer über vier Quadrate mit je 16 kleineren Kästchen. Unter dem Lichtmikroskop werden die Zellen in jeder dieser insgesamt 64 Quadrate kumuliert und durch vier dividiert. Dadurch erhält man einen Mittelwert der vier Hauptquadrate. Diese mittlere Zellzahl wird nun mit dem Faktor Zehn multipliziert und es ergibt sich die Zellzahl pro Mikroliter. Der
Material und Methoden
Faktor Zehn ergibt sich aus der Fläche von einem Quadratmillimeter für jedes der vier Hauptquadrate und der Kammerhöhe von 0,1mm. Daraus folgt ein Volumen von 0,1 l pro Quadrat.
Da nun die Zellzahl bekannt war, konnte eine vorher ebenfalls gelatinierte 24- well-Platte mit 50000 Zellen pro Well beimpft werden. Nach Zugabe von je 1ml endothelialem Nährmedium, wurden die Zellen für zwei Tage bei 37°C inkubiert.
Abbildung 6 HUVEC unter dem Mikroskop
2.2.8.2 Versuchsdurchführung mit Acetylcholinchlorid
Das Wachstum der HUVEC in den 24-well-Platten wurde lichtmikroskopisch verfolgt und ab einer Konfluenz von 80-90% der Versuch gestartet. Hierzu galt es zunächst, das verbrauchte endotheliale Nährmedium gegen frisches Medium zu ersetzen und anschließend die einzelnen Wells mit verschiedenen Acetylcholinchlorid-Konzentrationen zu beimpfen.
Material und Methoden
Getestet wurden die Konzentrationen von 0,1 M bis 100 M aufsteigend in Zehnerpotenzen mit drei unterschiedlichen Inkubationszeiten (30, 60, 120 Minuten). Des Weiteren wurden je Inkubationszeit auch zwei unbehandelte Wells vorbereitet. Diesem Schema entsprechend, wurden die HUVEC in den 24-well- Platten dann bei 37°C inkubiert.
Nach Ablauf der jeweiligen Inkubationszeit wurde das Acetylcholin-belastete Nährmedium entfernt, die Zellen mit Hanks BSS (buffered salt solution) gespült und mit neuem Nährmedium überschichtet.
Im Anschluss erfolgte die Zugabe von Interleukin 1-beta (finale Konzentration:
50ng/ml) in je zwei Wells pro getesteter Acetylcholinkonzentration sowie den unbehandelten Zellen. Somit verblieben von den ursprünglich vier Wells je Konzentration und Inkubationszeit noch je zwei als unstimulierte Proben. Die HUVEC wurden nun für sechs Stunden bei 37°C inkubiert.
Nach Ablauf dieser sechsstündigen Inkubation wurden das Nährmedium entfernt, die Zellen gespült und mit Antikörpern gegen CD54 (ICAM1) sowie CD106 (VCAM) beimpft. Beide Antikörper waren mit Fluoreszenzfarbstoffen (FITC bzw. PE) konjugiert. Die Inkubation fand für 45 Minuten bei 4°C statt. Danach wurden die Zellen wiederum mehrfach gespült um überschüssige Antikörper abzuwaschen.
Im Anschluss wurden die Zellen via Trypsinierung von den 24-well-Platten gelöst, in 5ml FACS-Röhrchen überführt und umgehend bei 400x g und 4°C für 5 Minuten zentrifugiert.
Der Überstand wurde verworfen und das Zellpellet in Hankss Buffered Salt- Solution resuspendiert. Zum Schluss erfolgte die durchflusszytometrische Messung der Oberflächen-expression von CD54 sowie CD106 auf den endothelialen Zellen.