Erhöhung der Antigen-Selektivität von T-Zellen durch Koexpression chimärer Antigen- Rezeptoren unterschiedlicher Spezifität
204
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
(42)
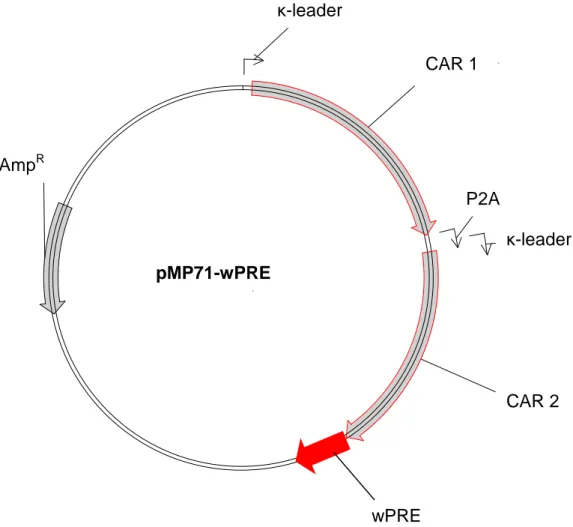
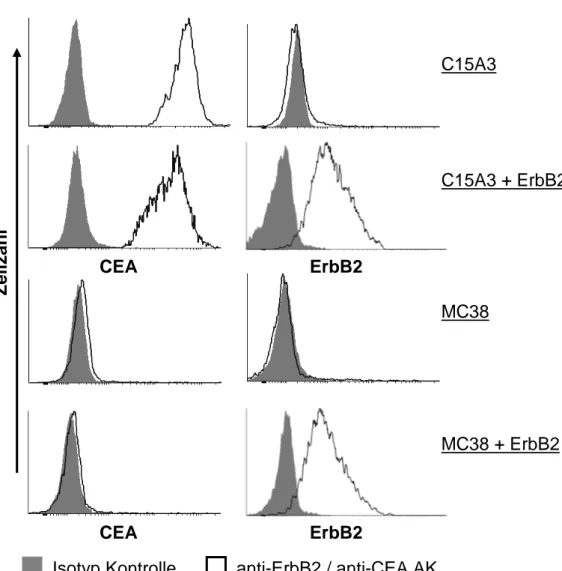
Abbildung
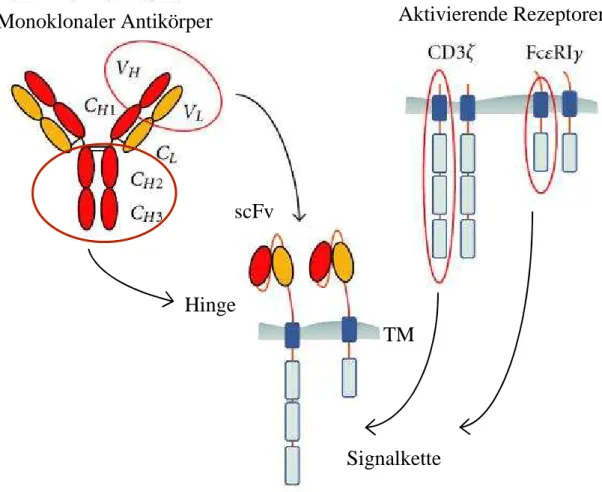



+7




ÄHNLICHE DOKUMENTE
Selbst wenn der TCR-3825 als TCR für den adoptiven T-Zell-Transfer ausscheiden sollte, so können die TCR-3825 transgenen T-Zellen als Testsystem für
Nach Stimulation der Co-Kulturen durch Lipopolysaccharide zeigte sich die Sekretion von IL-6 aus postoperativ gewonnenen Makrophagen in Co-Kultur mit postoperativ
Generell konnte daher in den hier durchgeführten Experimenten ausgeschlossen werden, dass lösliche Faktoren hauptverantwortlich für die durch Infektion verminderte IL-2-Produktion
Zur Be- urteilung der Abtötung dieser Zellen durch anti-TSHR CAR-T-Zellen wird die Minderung der Zielzellen in Ko-Kultur unter dem Mikroskop, dem Fluoreszenz- mikroskop
In dieser Stimulationsphase wurden auch Ansätze mit Cytostim (Positivkontrolle) und ohne Antigen (Negativkontrolle) mitgeführt. Dabei wurden die Proben mit dem pp65
In der vorgestellten Arbeit konnte gezeigt werden, dass die Sekretion von IL-6, IL-1β und IL-23 durch MoLC nach der kombinierten Stimulation des TLR2, 4 und 9
(3) Ein endogenes Antigen wie das transgene Ovalbumin führt nach der Aufnahme und Pro- zessierung durch LSEC, KC oder DC der Leber NICHT zu einer nennenswerten Aktivie- rung
Allerdings induzieren TCR modifizierte T-Zellen auch ohne CD28 Kostimulation eine höhere T-Zell Aktivierung und haben eine niedrigere Aktivierungs- schwelle als die
