Combined STM/AFM with functionalized tips applied to individual molecules
170
0
0
Volltext
(2)
(3)
(4)(5)
(6)
(7)
(8)(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
Abbildung
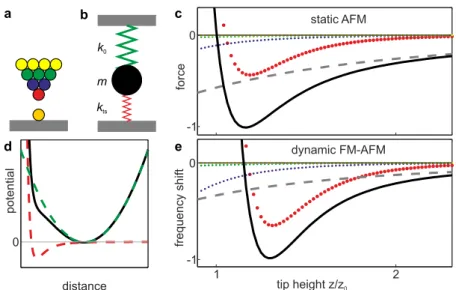
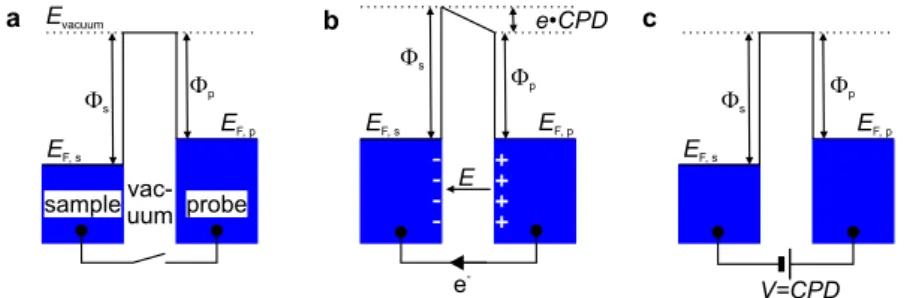
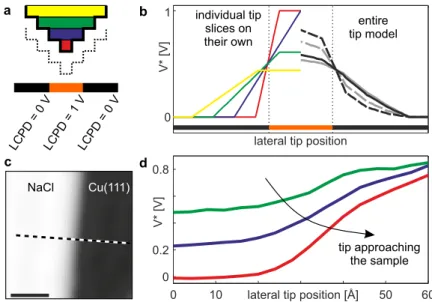

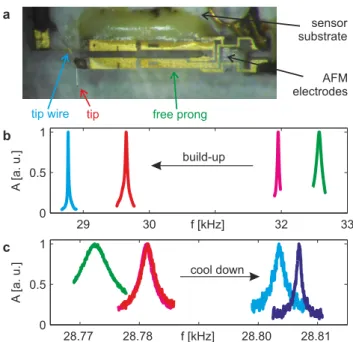
+7


ÄHNLICHE DOKUMENTE
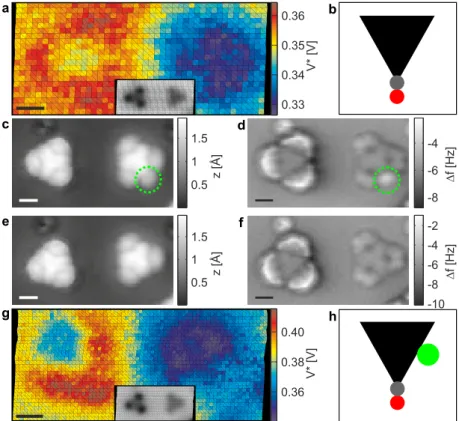
In this regime, KPFS maps have shown a contrast inversion [10] which hints that they may not reflect the charge distribution anymore [10].. Here we study
Here we show by a combination of angle-resolved photoemission (ARPES), scanning tunneling microscopy (STM), and atomic force microscopy (AFM) that the protection mechanism of
Assuming a linear relationship between lateral distortion and force, atomic force microscopy images could be deskewed for different substrate systems.. DOI:
The direct correspondence between the experimental STM images and the molecular orbitals of the free molecule clearly demonstrates that the electronic decoupling pro- vided by
Left: A magnetic atom that is directly evaporated on a metal surface is subject to strong interactions with the substrate, mainly electron scattering leading to a suppression of
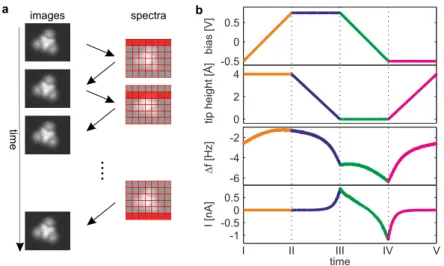
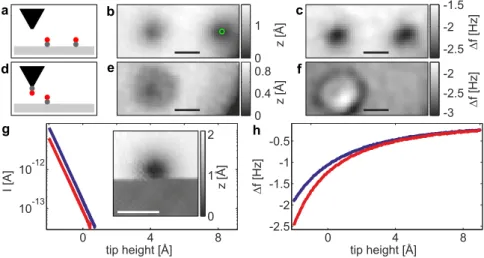
Combined Scanning Tunneling and Atomic Force Microscopy and Spectroscopy on Molecular Nanostructures
Scanning probe methods like scanning tunneling microscopy STM and atomic force microscopy AFM are the tools of choice for investigating the fundamental electronic, magnetic
While in the past zero-bias anomalies have usually been interpreted as Kondo resonance in the strong anitferro- magnetic (AFM) coupling regime, our analysis of the temperature
It is interesting to note that the corner atoms of a silicon cluster which is bounded by (111) planes must expose two dangling bonds per corner atom, unless one of the (111) planes
