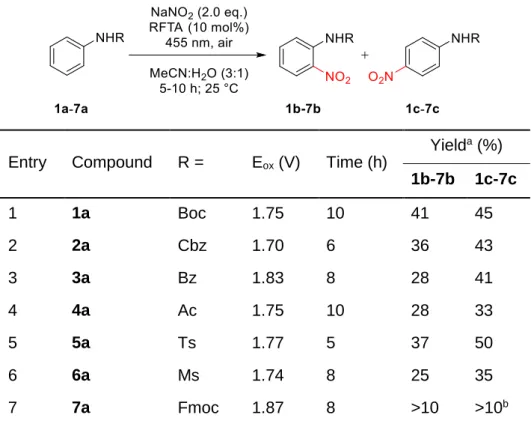
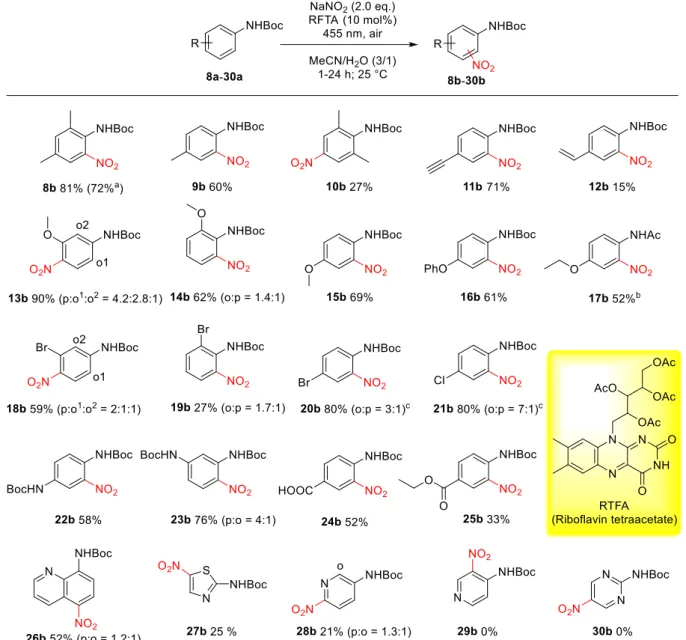
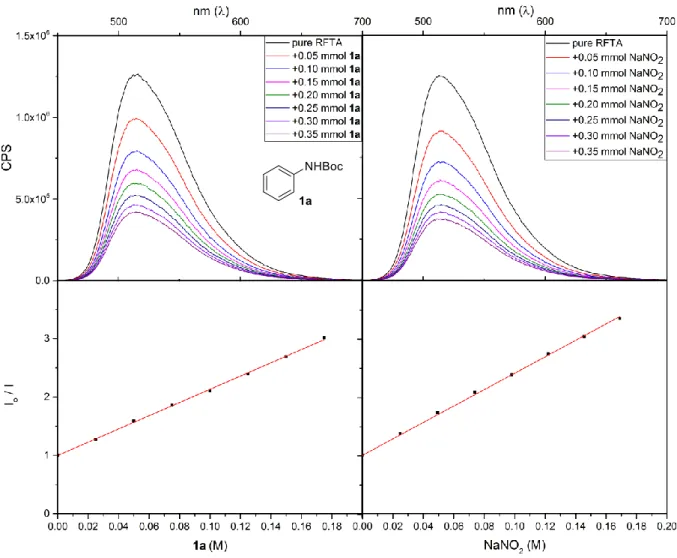
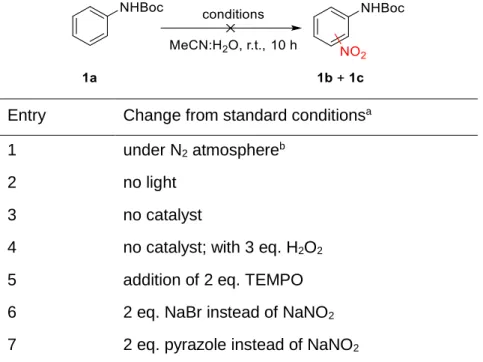
Functionalization of sp
160
0
0
Volltext
(2)(3)
(6)(7)
(8)
(9)
(10)
(11)
(12)
(13)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(29)
(30)
(31)
(32)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
Abbildung
+7
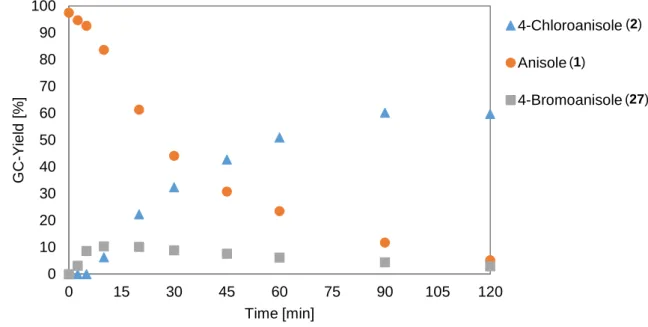
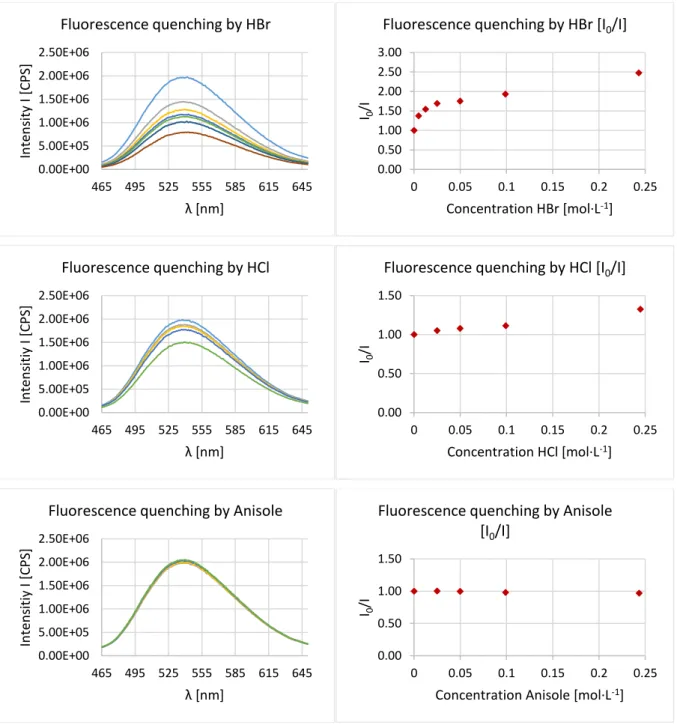
ÄHNLICHE DOKUMENTE
In this paper we have extended the threat model for human identification protocols by assuming a stronger attacker who can predict random decisions made by the human. We have stated
In the long term, Russia’s stance in the conflict with Ukraine will reinforce the viewpoint shared by some in the German government that the Kremlin’s ruling elite has a
This is one way in which to avoid the type of situation described by Magay (1984: 224): "Whether traditional human methods or computer-aided procedures are used, help must be
In general, our chemical analyses of these previously unstudied scent volatiles of Aspavia brunna (Heteroptera: pentatomidae) revealed some interesting divergence from those of
6.1 Materials supply: If the buyer supplies the seller with materials needed for contract per- formance, these shall remain the property of the buyer.. They have to be designated as
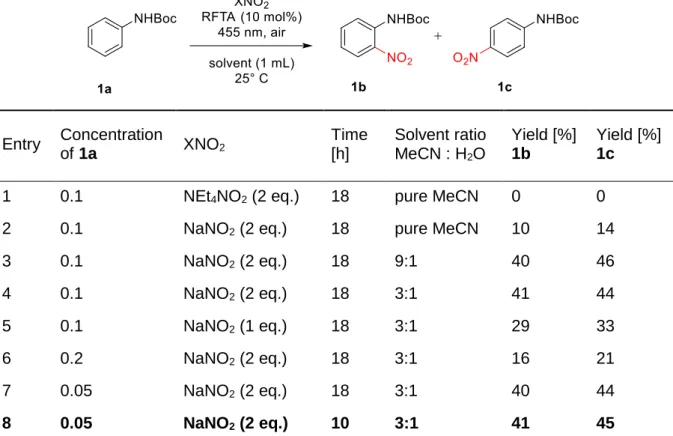
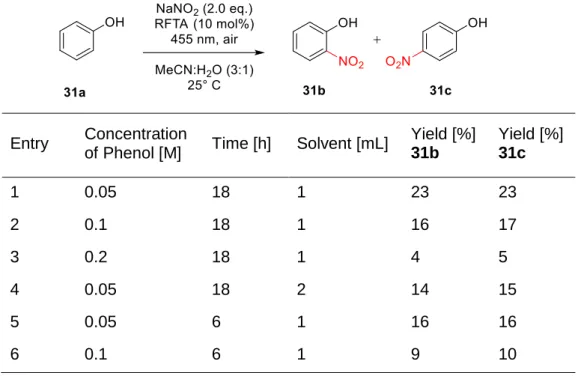
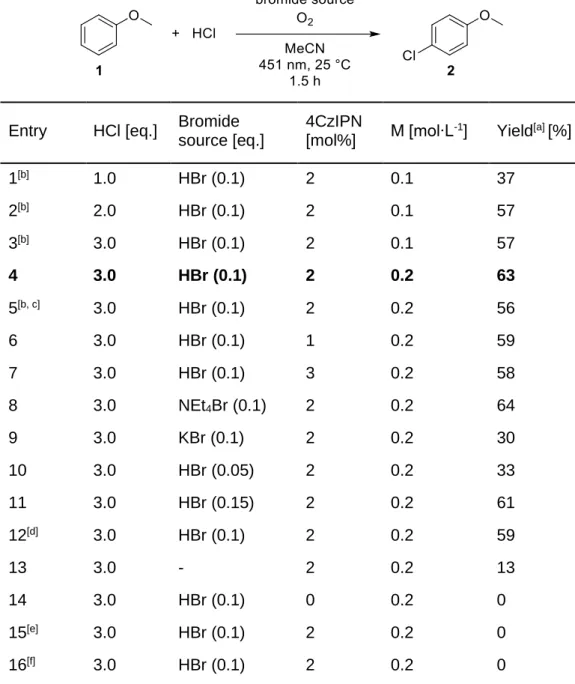
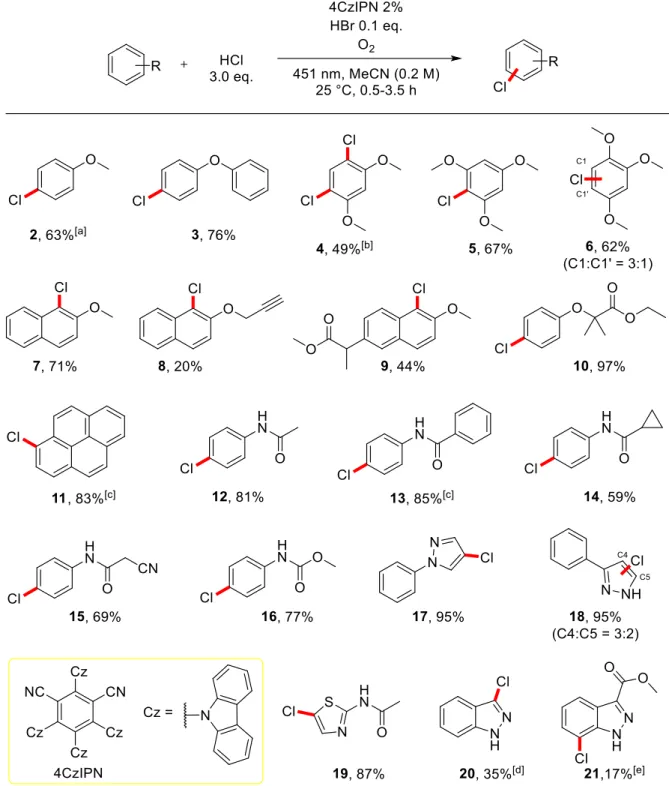
The filtrate was evap- orated and the crude product purified by chromatography (silica gel, hexane/ethyl acetate 2:1) to afford 6 (3.95 g, 82 %).. The reaction mixture was diluted
The coordination sphere of the silver atoms is further complemented by an ethanol molecule which is also engaged in hydrogen bonding with one of the sulfonate oxygen atoms..
I refer to my letter dated 6 June 2019, in which I informed you that I would convene an informal meeting of the General Assembly on “Combatting Antisemitism and Other Forms of
