Research Collection
Journal Article
The Formation of ThRh Hydride and the Synthesis of Methane by its Reaction with CO
Author(s):
Berner, H.; Oesterreicher, H.; Ensslen, K.; Schlapbach, L.; Schlapbach, L.
Publication Date:
1982
Permanent Link:
https://doi.org/10.3929/ethz-b-000423131
Originally published in:
Zeitschrift für physikalische Chemie 132(1), http://doi.org/10.1524/zpch.1982.132.1.075
Rights / License:
In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use.
ETH Library
Zeitschrift furPhysikalischeChemieNeueFolge,Bd. 132,S. 75-84(1982)
© byAkademischeVerlagsgesellschaft, Wiesbaden 1982
The Formation of ThRh Hydride and the Synthesis
of Methane by its Reaction with CO
By
H. Berner, H.
Oesterreicher1,
K. EnsslenFakultat fiirPhysik,UniversitatKonstanz, D-7750Konstanz,WestGermany and
L.
Schlapbach
Laboratorium fiirFestkorperphysik,ETH, CH-8093Zurich,Switzerland
(ReceivedJuly26, 1982)
Metalhydride / Surfacesegregation/ Catalysis /Methanation
Thecompound ThRh forms ThRhH3A under 1bar H2 pressure at room temperature.
Thermodynamic analysis yieldsAH°=
—
58.7kJ/mole H2and AS"=
—
128J/K moleH2.A
methanation reaction is observed whenThRhH3[isexposedtoCO. Apreferentialreaction of CO with thehydrogenstored in the metalhydriderather than with thehydrogenin the ambient gas phase is shown by use ofhydrogen-isotopes. XPS experiments ofThRh and ThRhHz
demonstrate surfacesegregationintoTh02 and Rhclusters,whenexposedto02 orCO.
Die intermetallischeVerbindungThRhbildetbeieinemH2-Gleichgewichtsdruckvon1 bar
undZimmertemperaturdasHydrid ThRhH3dessen BildungsenthalpieAH"= —58.7kJ/mol H2 und Bildungsentropie AS°=-128J/K mol H2 betragt. Wird ThRhH3, einer CO-
Atmosphareausgesetzt,ist dieEntstehungvonMethan nachzuweisen. Mit HilfevonWasser-
stoffisotopen wird gezeigt, daB bei dieser Reaktion der Wasserstoff, der im Metallhydrid gespeichertist,gegeniiberdem WasserstoffausderGasphase bevorzugt eingebautwird. XPS- UntersuchungenvonThRh undThRhHz zeigenbeiZugabevon02bzw. CO eine Oberflachen- zersetzung,bei derTh02- und Rh-Teilchenentstehen.
Introduction
Metals andintermetallic
compounds consisting
ofrareearthsoractinidesincombinationwith transition metalscanabsorb
large
amountsofhydrogen.
1GuestProfessor, permanentaddressatUC-SanDiego,La Jolla where work issupported by
agrantfrom theDepartmentofEnergy, BasicEnergySciences
Onestep of thisprocessis the dissociation of the
H2
moleculeatthe surface.As shown
by
theexample
ofLaNi5 [1]
asegregation
of therareearth element atthe surface takesplace, binding
theoxidizing impurities
andkeeping
thetransition metal in the metallicstate. Inthiswaysitesare
created,
whichareabletodissociate molecules. On the other hand these
compounds
candeliverlarge quantities
ofatomichydrogen
from the bulk.They
therefore should begood
candidates ascatalysts
forhydrogenation
reactions.Wallaceetal.
[2
—4] investigated
variouscompounds
ofthistypeintheirunhydrided
statewithregard
totheircatalytic activity
in theNH3
andCH4 synthesis. They found,
that under thesynthesis
gas CO andH2
the com-pounds decompose
intocrystallites consisting
of the transition metal and ofanoxide of therareearth/actinide
which showedatleastinone casehigher activity
than aconventional oxidesupported catalyst
of thesame elements.Soga
etal.[5]
studied thehydrogenation
ofethylene
overLaNi5
andfound,
that the rate of
hydrogenation
over thehydride
waslarger
than over theunhydrided
material. Oesterreicheretal.[6]
showed thecatalytic
formationofwater, when
hydrides
ofintermetalliccompounds
wereexposed
toair.Inthis workwestudied the
CH4 synthesis
from carbon monoxideoverThRh
hydride.
We choose ThRhas amodelcompound type
"rareearth oractinide/transition
metal" for several reasons:a)
the material isexpected
to form ahydride,
which is even stable attemperatures
of 200-300°C.
b)
Rhodium could becomecatalytically
active in clustersize on thesurface,
if theexpected segregation
ofThorium occurs.c)
Thorium isreported [4]
toforma moreactivesupportthanLa,UorZr.In addition to a
study
ofthehydriding
behaviour of ThRh and the reactionof ThRhhydride
withCO,
XPSwasused tohelp understanding
ofthe surface processes.
Experimental
details and results1. Material
ThRhwasprepared by meltingthecomponentstogetheron awatercooledcopperhearth of
anarcfurnace under argonatmosphere.The buttonwasflippedand remelted several timesto
improve homogeneity.
The orthorhombiccrystalstructure[7]wasverifiedby analysisofX-ray powder diagrams (Guiniercamera,Cu-A'aradiation).Therewasalso evidence ofTh02presence, butnotof Thor
Rh.
2. Hydriding
ThRhwashydridedin thereactorofahigh-pressurestainless steelapparatuswith various reservoirs of known volume and facilitiestoworkatdifferentpressure ranges.Thetemperature
was monitored by a NiCr —Ni thermocouple in the reactor bed. For the variation of the temperature,anexternal heaterwasused.
The Formation of ThRh Hydrideand theSynthesisof Methane 77
ThesamplewasexposedtoH2 gas(99.999%), additionally purifiedfrom02and H20 by chemisorption and molecular sieve filtration, respectively, accomplished by passing the gas
through a commercially availablecartridge (Oxisorb, Messer Griessheim). The amount of
absorbedhydrogenwascalculated from thepressuredrop.The first reaction withH2(28 bar)
occurredspontaneouslyatroomtemperaturewithoutpreviousactivation.Atthistemperature and1barequilibrium H2pressurethehydride ThRhH3,is formed. Thepressure-composition isotherms,shown inFig.1weremeasured for fourtemperaturesby monitoringtheequilibrium H2pressurestartingfromapoint,whereThRh isinthehydride phaseandpumpingoutknown quantitiesofhydrogengas. Threeof thesedesorptionisotherms showplateaus,associatedwith
the transformation of thehydride (/?) phaseinto the solid solution(a)phase. Thermodynamic parametersderived fromalogarithmic plotofplateaupressurevs. reciprocaltemperatureare AH"= -58.7kJ/mole H2 and AS"= -128J/K molr H2.The X-ray pattern of the ThRh
hydridecouldnotbe indexedby enlargingthe orthorhombic cell ofThRh,norbythepresenceof
ThH2orTh4H15.
3.
Catalysis
3.1.
Apparatus
The
catalysis experiments
wereperformed
inthehydriding apparatus by
useof
corresponding parts
of itas asingle
passreactor(volume
5cm3)
or as aclosed
system.
This contained several chambers forfilling
in knownamountsof different gases. In addition to CO
(99.97%), H2 (99.999%)
andD2 (99.4 %) required
for theCH4 synthesis,
Ar(99.997 %)
wasusedas areferencegastocheck
changes
inthehomogeneity
ofthegasmixture and in thepartial
pressures ofthe
components.
A
quadrupole
massspectrometer (Balzers, QMS)
builttogether
with aseparate high
vacuum station was used. Thecomponents
of the gas werequantitatively analysed by considering
the known ratios ofsingly
anddoubly
ionized and
fragmentary
molecules andby
alsoincluding
a factor for theprobability
ofionization,
which isdifferentfordifferentgases[8,9].
Since thepeak
atthe atomicmassunity (amu)
16iscomposed
notonly
ofCH4
but alsoof CO and
H20,
thepeak
atamu15(CH3-fragment)
wasusedtodetect thepresence of
CH4.
3.2. Procedure
AThRh
piece
of about 0.75gwashydrided
atroomtemperature,
cooledto
—
50 °C and after
removing
thegaseoushydrogen, exposed
toCO and Ar.Then thereactorwasheatedto100°Cand
part
ofthehydrogen
inthehydride
allowedtodesorb. This
temperature
washeld for about 12h whilethegaseswere circulated in the closed
system,
drivenby convection,
to reachequilibrium
andhomogeneity.
This was confirmedby observing
constanttotalpressureand
obtaining
thesameratios of thepartial
pressuresasthoseratios determined from the filled in
quantities.
Either the closedsystemwas usedduring
thewholeexperiment
orthereactorwasswitchedovertoasingle
passreactorand thegases
passed
overthe ThRhhydride
ata constantfeedrate of1 ml
gas/min (standard pressure).
This ratewas limitedby
the massspectrometer,
which was run at 1 •10~4mbar.
At the end of eachsingle experiment
the reactor wasevacuated,
the gas furtheranalysed by
massspectrometry
and thehydride
wasoutgassed
at200 °C for about 1h. Thisprocedure
wasrepeated
several times with the same ThRhsample.
3.3. Results
In all
experiments
withexposing
ThRhhydride
toCO, CH4
wastheonly
detectable
product.
The presence ofH20
wasnot detected eitherduring
theexperiment
orduring
evacuation at the end of theexperiment.
Further aprogressive
decrease ofthehydrogen
storagecapacity
was observed. Thisfact, together
with the absence of oxygencontaining
gaseousproducts
andtherecordedpressure decrease
during CH4 synthesis suggest,
that the ThRhsample
became more and more oxidized.Ineach successive
experiment
the time and thetemperature,
atwhich thebeginning
of theCH4 synthesis
could bedetected,
was lower than in theprevious experiment.
We took the detection of1%
ofCH4
relativetothe totalThe Formation of ThRhHydrideand theSynthesisofMethane 79 300-
L,
200-•- 100- 1000,
t/1
<
UJ LU o =>
en -J LUD_
Ui
50
H
vH2
0.75 0.60 QMS
-I
0.150
o
£
-I
0.30ffi
<i i i—i T—i-—r
0 10 20 30 40 50 60 70 80 TIME (min)
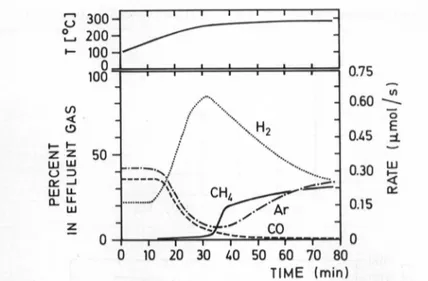
Fig.2. Rate ofCH4formationfrom COoverThRhHzduringincreaseof thetemperature.H2
evolves from thehydride.Ar isusedas a"feed rate" reference
amount of the
analysed
gas as a measure for the onset. In thevirgin hydrogenation cycle
of ThRh theonsetis found after12hat100"Cplus
3.5 hat200°C. Inthe8th
catalysis cycle
itcouldbealready
seenafter12 hat95°C.The total pressure was 1.1 and 1.2
bar, respectively.
Westudiedmoredetailed the influence of the
desorption
of thehydrogen
from the metal
hydride
ontheCH4
formation.InFig.
2thegasmixture andtemperature
course in asingle
passexperiment
are shown. Atemperature
increase leadstoincreasedhydrogen desorption, initiating instantaneously
ahigher
rateofCH4
formationcompared
with the fraction ofargon,which isareference of thefeedrate
during desorption.
Toshow thepreference
in themethane
synthesis
between thehydrogen
evolved from thehydride
and theambient
H2
gas,anisotope experiment
wascarriedout. ThRhwashydrided
and after
removing
most of the gaseoushydrogen, exposed
to carbonmonoxide and deuterium in the closed
system. During
evacuation of the reactorthemassspectrum
showedpeaks
atamu2,
3and4in the ratio 7:6:8arising
fromH2,
HD andD2
andpeaks
atamu12to20,
which refertoseveral C—
H
—
D
compounds
listedinTable1.Although
the presence ofmoreD2
gas than
H2
gasisevident, CH4
andCH3D
are dominant overCD4.
Fromthese results we
conclude,
that informing methane, hydrogen
from thehydride
ispreferred
overhydrogen
from thegasphase.
After7
experiments
with CO and 10hydriding cycles,
the ThRhpiece
haddisintegrated
topowder
and had increased itsweight by 7%. X-ray
diffraction
analysis
showedTh02, ThRh3
and Rh. Thissample
was thenexposed
tooxygenat200°Cfor12 handthe resultofthesubsequent catalysis
Table1. Distributionofreactionproducts ofCO andD2 overThRhHzat171°C
_J_I_I_I_
CH*
CH3D CH2D2 CHD3 CD<0 50 100 150 200 250 300
TIME (min)
Fig.3. Formation ofCH4andC02from CO andH2 overthe oxidized ThRhsample during increaseof thetemperature.H20is alsoformed(seetext).Aris usedas a"feed rate" reference
experiment
is shown inFig.
3.Hydrogen
was admitted inthe gaseousstate, because thematerialdidnottakeupanymorehydrogen.
Thedecreaseof COcombinedwiththe increase in
CH4 implies,
thatCH4
is formed from CO and not fromC02,
which was alsoproduced.
Water was detected inlarger
amounts
during
evacuation and therefore not marked in thefigure.
TheX-ray
diffractionanalysis
showed that the material consists ofTh02
and Rh.The last
experiment
carried out without the ThRhsample
or itsdecomposed products confirmed,
that noproducts
at reaction conditionswere formed
by experimental
artefacts.The Formation of ThRhHydrideand theSynthesisof Methane SI 4. Surface
analysis
The ThRh surfacewasstudied
by X-ray photoemission
spectroscopywitha
VG-Escalab-Spectrometer using Mg-Ka
radiation(hv
=1253.6eV;
Au4fV2
at83.9eV,
FWHM 1.2eV).
The ThRhwascleft underUHVconditions(2
—
4-
10~10mbar)
and afteranalysing
the clean surface thesamples
wereexposed
in situ toincreasing
doses of02
andCO, respectively.
Thephotoelectron
energy distribution curves of the core levels Th4/7/2i5/2,
Rh
3J5/2
3/2, O 1s1/2 and C 1s1/2
were measuredto evaluate the chemical state and the concentration ratios within the escapedepth
of thephoto-
electrons
(~20A).
The chemical state was determinedby comparison
toknown
spectra.
Theconcentration ratio wascalculated from theintegrated peak height
dividedby
theoretical cross sections[10].
In
Fig.
4thespectra
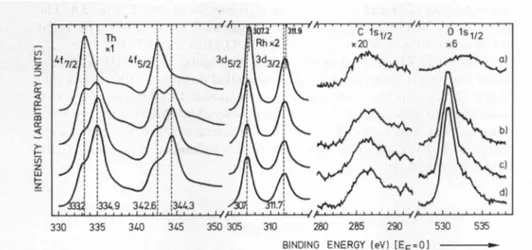
ofcleft ThRh before and afteroxygentreatmentareshown. The clean
sample
contains metallic Th[11,12]
and metallic Rh[13,14].
Withincreasing
amounts of oxygen,increasing intensity
of thechemically
shifted Th4/peaks
is observed. This isexplained by
formation ofTh02 [12]
inagreement
with the increase andposition
ofthe oxygenpeak.
TheRh
peak
shifts 0.2 eVtolowerbinding energies,
which could beexplained by
the formation of small Rh clusters with ahigh
amount oftop
surfaceatoms. The C
spectra
do notchange during
exposure to02.
The con-centration Th:Rh
changes
from 1.6:1 of the cleftsample
to2.7:1 after 100L02 (1
L= 1Langmuir
= 1•10~6Torr s).
Therefore02
exposure leads to enrichment of Th at the surface in the form ofTh02.
The Rh remains metallic.330 335 340 345 350 305 310 280 285 290 530 535 BINDING ENERGY (eV| [Ep=0]
-
Fig.4.XPSspectra of cleft ThRhexposedto02 at20 C:(a)cleftsample,Th:Rh=1.6:1;
(b)exposedtotL02,Th:Rh=2.1:1;(c)exposedto10LO2,Th:Rh=2.5:1;(d) exposedto
100L02,Th:Rh=2.7:1
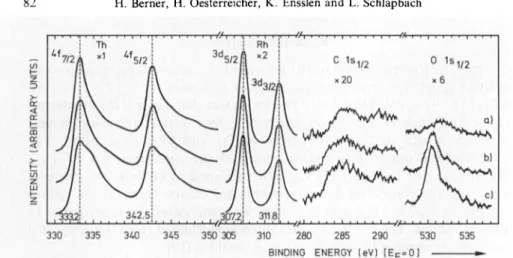
Fig.5. XPSspectraof cleft ThRhexposedtoCOat200 C:(a)cleftsample,Th:Rh=1.5:1;
(b) exposed to10LCO,Th:Rh= 1.6:1;
(c)
exposed to1 000 L CO,Th:Rh=1.8:1Further the
spectra
of ThRhwererecorded before and after COexposure atroomtemperature
andat200° C. Some of themareshown inFig.
5. Thesame effect
—
enrichment of Th as
ThOz
and metallic Rh—
as after
02
exposure isseen.
However,
ittakes 1000LCOtoproduce
thesameamountofTh02
as withonly
1L02.
The Cspectra
show new small contributions atenergies
between 282 and 285eV, especially
at200°C,
which couldoriginate
fromdissociatedCO
[15].
The ratio C: O howeverchanges
from1 :0.5 for the cleftsample
to 1 :1.2 after 1000 LCO,
sothat notall of the oxygen canbe derived from dissociated CO. Toanalyse
theamount ofsurfacecoverageby
oxygen,which diffuses from the bulktothe
surface,
wefollowed the surface concentration ofO and C ofafreshly
cleftsample
at200 °C over2h. Thecarbon
peak
didnotincreaseatall.Theoxygenpeak
increased40%
overthatin the clean
surface,
where the(Th
+Rh)
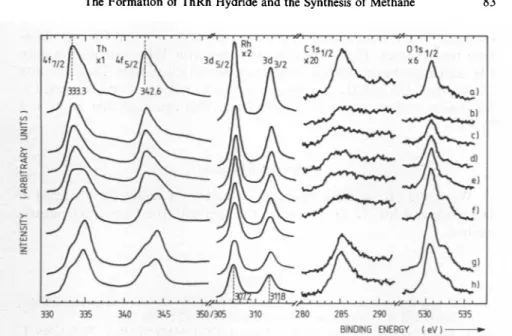
:0 ratio was 3.2:1.The cleftThRhwas
hydrided
inaseparate
chamber oftheUHV-system
at20 bar
starting
atroomtemperature
and cooled downto —100° Cto avoid strongH2 desorption.
Thehydride
wasexposed
to CO and afterwards warmeduptoremoveallhydrogen.
Thepowdered hydride
had the lowest of allmeasuredcontentsofimpurities
C and Oas seeninFig.
6. CO exposure of thatpartially hydrided sample
showsnofurther differenceascompared
totheunhydrided
material.Discussion
The derived
thermodynamic properties
of ThRhhydride,
AH0 =—
58.7
kJ/mole H2
and AS°= —128J/K
•moleH2,
arecomparable
tothoseof similar
hydrides
such as ThNi andThCo,
which arereported
in theThe Formation of ThRhHydrideand theSynthesisof Methane 83
330 335 340 345 350/305 310 280 265 290 530 535 BINDING ENERGY IeV)-*>
Fig. 6. XPSspectra of cleft ThRh and ofThRhHz exposedtoCO:(a)cleftsampleat20°C, Th:Rh=1.6:1; (b) hydrided, cooled to -100°C, Th:Rh=1.4:1; (c) hydride exposed to
10 LCO,Th:Rh=1.5:1;(d)hydride exposedto100 LCO,Th:Rh=1.5:1;(c)hydrideex- posed to 1000 L CO, Th:Rh=1.6:1;(/) hydride exposed to10000 L CO, Th: Rh= 1.8:1;
(g) hydride warmed up to 20 C, Th:Rh=2.5:1; (h) completely dehydrided,
Th:Rh=2.4:1
literature
[16],
Thus ThRhhydride
is stableenough
to be handled withoutstrong desorption
attemperatures
up to 200 °C. Thistemperature
rangeisnecessary to
study CH4 synthesis
from CO over the metalhydride.
Withthe enrichmentof Thas
Th02
onthesurfaceand the formation of metallicRh,
whenexposed
to02,
ThRh has theexpected
surfaceproperties anticipated
from similarexperiments
withLaNi5 [1],
where oxidized La and metallic Ni arefound in the ratio 1 :1 on the surface.When ThRh
hydride
isexposed
toCO, CH4
is formed belowtempera-
tures of 200°C. The
hydrogen evolving
from thehydride
ispreferred
informing CH4, compared
tothehydrogen
in the gasphase,
as shown intheisotope experiment.
This fact makesevident that theorigin
andstate of thehydrogen
isveryimportant.
Thispreference
appearstobe causedby
thelarge supply
of atomichydrogen by
thehydride.
In addition newclean,
i.e.catalytically
active surfaces may be createdduring hydriding
anddehydriding
of the intermetallic
compound by progressive disintegration
orsegregation.
However the oxygen of the carbon monoxide is used
subsequently
foroxidation of
Th, although
a"reducing atmosphere"
is formedby dehydrid- ing.
But if theCH4 synthesis
from CO would workonly
on the basis ofoxidation of
Th,
thecompletely
oxidizedsample
wouldnotshow anyCH4
synthesis.
This isnotthecase. TheX-ray
patternshows,
that Rhcrystallites
have been formed. These cristallites
together
withTh02
canalsoproduce CH4
andmight
be consideredasthoriasupported
Rhcatalyst.
The formation ofCH4
from CO andH2
overtheseverely by
CO oxidizedsample
startedatthe lowest
temperature
of allexperiments.
Thisextremely
low value was95°C.
Acknowledgements
We would like tothank Professor E. Bucher for
support
and interest in this work and Mr. G. G. Baumann forhelp
with the massspectrometric analysis.
References
1. H. C.Siegmann,L.Schlapbach, Phys.Rev. Lett. 40(1978)972.
-
L.Schlapbach,A.Seiler,
F. Stucki and H. C. Siegmann,J. Less-Common Met. 73(1980) 145.
2. T. Takeshita,W. E. Wallace and R. S. Craig,J. Catal. 44(1976)236.
-
V. T.Coon,T.
Takeshita,W. E. Wallace and R. S.Craig,J. Phys.Chem.80(1976)1878.
3. H. Imamura and W. E.Wallace,J. Catal. 65(1980)127.
4. A. Elattar, W. E. Wallace and R. S. Craig,Adv. Chem. 178(1979)7.
5. K. Soga, H. Imamura and S.Ikeda,J.Phys. Chem. 81(1977) 1762.
6. H.Oesterreicher,K. Ensslen and E.Bucher, Appl.Phys.22(1980)303.
-
H. Oesterreicher and F.Spada,Mater. Res. Bull. 15(1980)477.
7. Shunk,Constitution ofBinary Alloys,SupplementNr. 2 McGraw-Hill,New York 1969.
8. Index of MassSpectral Data,AMD11,ASTM.
9. Balzers,PhysikalischeGrundlagenzurParlialdruckanalyse (14.9.1973).
10. J. H.Scofield,J. ElectronSpectrosc.8(1976)129.
11. S.B. Nornesand R. G. Meisenheimer,Surf. Sci. 88(1979) 191.
12. J. C.Fuggle,A. F.Burr,L. M.Watson,D. J. Fabian and W.Lang,J.Phys.F:4(1974)335.
13. R.Nyholmand N. Martensson,J. Phys.C: 13(1980) L279.
14. T. L.Barr,J.Phys. Chem. 82(1978) 1801.
15. J. C. Fuggle,in: Handbookof X-rayand ultravioletphotoelectronspectroscopy(D.Briggs Ed.),Heyden (1977).
16. H. H. vanMai, Proefschrift(1976).