Synthesis of mannose extended N- and O-glycopeptides for antibody generation and studies of protein-carbohydrate interactions
355
0
0
Volltext
(2)(3)
(4)(5)
(6)(7)
(8)(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
(42)
(43)
(44)
(45)
(46)
(47)
(48)
(49)
(50)
(51)
(52)
(53)
Abbildung

+7
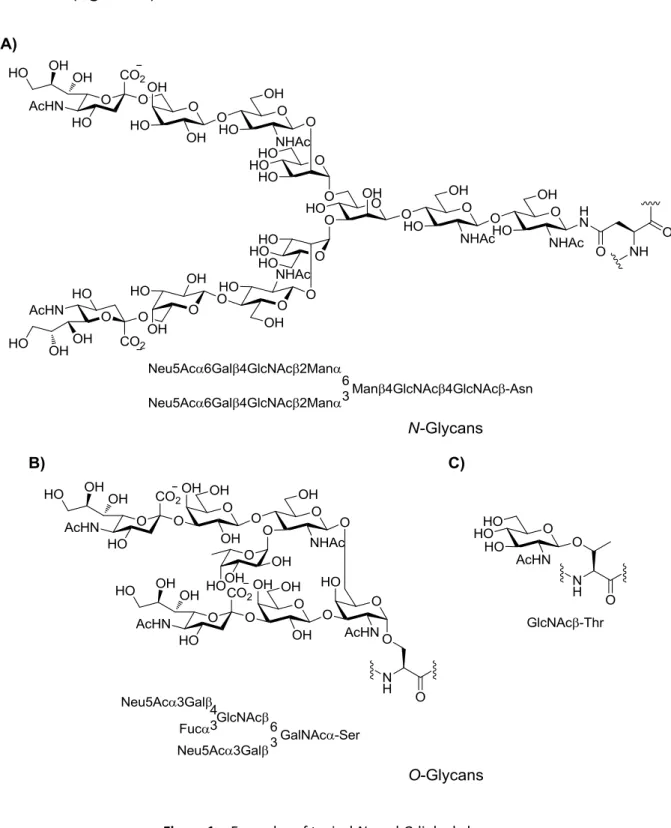
![Figure 9. Homogeneous native N-glycan structures. [123]](https://thumb-eu.123doks.com/thumbv2/1library_info/3632523.1502276/50.892.220.672.418.805/figure-homogeneous-native-n-glycan-structures.webp)
ÄHNLICHE DOKUMENTE
Key words: Potassium, Hydroxylamine, Aggregate, Bond Cleavage,
Two unnatural α -amino acid esters were prepared in good yields via phase transfer catalyzed Michael addition of ethyl N-acetylaminocyanoacetate to chalcone and benzalketone.. For
Two unnatural α -amino acid esters were prepared in good yields via phase transfer catalyzed Michael addition of ethyl N-acetylaminocyanoacetate to chalcone and benzalketone.. For
~ 4.32, corresponding to a randomly generated se- quence (1/20 probability of finding any one of the 20 amino acids at any given site). This level of complexity is never realized
[r]
The second group II in which a guanidine is incorporated instead of the second bis(Zn(II)-cyclen) triazine complex 16 should have a high affinity to peptides containing
In the heart conditional Hccs KO model, parts of the Hccs gene are specifically knocked out in cardiomyocytes from 7.5 dpc onwards applying Cre/locus of X-over P1 (loxP).. In
The criteria used to describe the influence of the investigated amino acids on the nucleation of cal- cium carbonate are; (1) the slope of the linear increase during the
