Research Collection
Doctoral Thesis
The Surface and Bulk Molecular Structures of Polyacrylamide Hydrogels and Their Influence on Mechanical and Tribological Properties
Author(s):
Gombert, Yvonne Publication Date:
2020-06
Permanent Link:
https://doi.org/10.3929/ethz-b-000447788
Rights / License:
In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use.
ETH Library
DISS. ETH NO. 26912
THE SURFACE AND BULK MOLECULAR STRUCTURES OF POLYACRYLAMIDE HYDROGELS
AND THEIR INFLUENCE ON MECHANICAL AND TRIBOLOGICAL PROPERTIES
A thesis submitted to attain the degree of DOCTOR OF SCIENCES of ETH ZURICH
(Dr. sc. ETH Zurich)
presented by Yvonne Sabine Gombert
MSc ETH Materials Science, ETH Zürich born on 17.03.1986
citizen of Germany
accepted on the recommendation of Prof. Dr. Nicholas D. Spencer, examiner
Prof. Dr. Jan Vermant, co-examiner Prof. Dr. Alison C. Dunn, co-examiner
2020
when it came to being she said be tender and tough at once you need to be vulnerable to live fully but rough enough to survive it all
rupi kaur the sun and her flowers
i
Every journey is an adventure. And every adventure is a sequence of big and small, exciting and boring, happy and unhappy moments. My time as a doctoral student was such a journey. I lived through good days and bad days, experienced fun moments to enjoy and tough challenges to survive. I feel that my PhD journey has shaped me more than any other journey in my life to date. What made these years worth living is the people that travelled with me and the people I met along the way.
Having reached the end of this journey, I would like to thank all those who enriched my life and accompanied me during my intense time here at ETH.
The person who started this adventure with me is Nic Spencer. Thank you, Nic, for giving me the opportunity to undertake this journey. You also provided me many more (flight) tickets to big and small adventures along the way. Together, I counted, we traveled to ten countries in five years. Traveling, besides science, I am convinced is a passion we both share. Other than the traveling, I am deeply grateful for your continued trust in my work, which has always given me the freedom for scientific as well as personal growth.
Another professor who had a major influence on my doctoral studies, especially at the beginning, is Greg Sawyer. You welcomed me to your team & your laboratory with open arms and generously shared your expertise on hydrogels with me. Thank you, Greg, for making our work together such an exciting and valuable experience inside and outside the lab. I will always remember your unique lessons about fishing, shark catching, mudding, and your introduction to the American culture in form of BBQ's, BLT sandwiches, glazed doughnuts in dozens, and s'mores.
Directly connected to my Florida adventures are my friendships with Angela and
Acknowledgements
ii
Juan. Thank you both for guiding me through countless hours in the lab, for shared Gin & Tonics, a road trip all over Florida (Juan), hiking in Switzerland and the real Oktoberfest experience in Germany (Angela).
Moh. You became a loyal lunch mate during the second phase of my PhD. Your scientific assistance is as much appreciated as the friendship we share. Certainly, our expedition through Iran is one of the greatest adventures I have had in recent years. Thanks go to you and your family who made this possible for me.
Matthias – you are the one who voluntarily picked up the task of supervising me and directing my work (back) on track. I am grateful for your belief in the value of my work, which has always strengthened my motivation.
This one goes to the “former” troop of LSST E-floor lab and beyond: Thank you Vikrant, Matthias, Rebi, Josephine, Clement, Andrea, Fabiana, Shiva, Alok, Szymon, Manjesh, Moh, Ella, Cristiana, Federica, Philip, Chris (in random order) for an active and enjoyable lab environment with fun parties and outside-work activities. It was great while it lasted! In this context, I would also like to cherish the help of Wenqing, Tobi and Shiva. Although we did not work together every day, you were constant and reliable support in the sometimes-turbulent work days in the lab. Thank you.
Josephine. You took care of all administrative work connected to the many adventures throughout my PhD. You proved not only a dedicated and helpful coworker but also became a good friend with whom I enjoy spending time with. I could always count on you during the last years. Thank you for being part of my PhD time.
I thank Giovanni for always helping me immediately with any kind of technical challenge I faced during every day lab work. I am grateful for having experienced your reliable and calm assistance not only in the technical field of work life but also on the personal level during my PhD. Thank you for showing you cared and for putting a smile on my face when I expected it the least.
During my doctorate, I had the pleasure to work with the students Tobi, Elen, Lucile, Rachel and Fabrice. I thank you all for your contributions to my work and
iii
your trust in my supervision. In particular, I would like to thank Fabrice Roncoroni for our long-lasting and successful collaboration. I have enjoyed working with you.
At times, social life in a continuously changing and shrinking work environment proved to be unexpectedly rocky. On that note, I would like to thank all team members of Eric Dufresne’s research group for always welcoming me among them as a lab friend and the team members of Markus Niederberger’s research group for leisurely after-work beer evenings at Alumni Lounge.
Chapter 2. I thank Prof. Peter Walde for his guiding and continuous support with UV/vis measurements. Dr. Guido Panzarasa contributed with fruitful discussions on chemistry details of O2-exposed FRP of pAAm hydrogels. Thanks also go to Katrina Smith-Mannschott for her patient help with acoustic resonance spectroscopy and to Dr. Mohammad Divandari for his support with the synthesis of TEMED amine oxide.
Chapter 3. I would like to thank Dr. Antoni Sanchez-Ferrer for the X-Ray Scattering measurements and his helpful support with data interpretation.
Falk Lucas from ScopeM assisted with SEM sample preparation and Fabrice Roncoroni captured the SEM images. Thanks go to Dr. Robert Style for his valuable advice on hydrogel and rubber elasticity and to Prof. Dr. Qin Xu for assistance with relaxation measurements.
Chapter 4. I would like to express my gratitude to Dr. Thomas Geue for his assistance during and after the days of neutron reflectivity measurements at AMOR/PSI. Fabrice Roncoroni contributed with IR and indentation measurements in the frame of his master project and Dr. Matthias Dübner gave valuable input through fruitful discussions and the initiation of IR measurements. Dr. Rok Simič assisted with the PSI data acquisition and with indentation as well as friction measurements.
The knowledge about contact angle measurements of Dr. Doris Spori has proved to be extremely valuable throughout my doctoral studies. Thank you, Doris, for sharing your expertise with me.
iv
Life Beyond the Lab
Dear Sarah, you helped me value the positive in every stone along the way. With your help I have learned to turn a defeat into an enriching experience and to use them as personal growth opportunities. These lessons you taught me were perhaps the hardest, but certainly the most rewarding and probably most lasting of my PhD adventure. Thank you for guiding me into this empowering life approach.
Liebe Nicole, unsere Freundschaft hält jetzt schon 15 Jahre obwohl unser Alltag in all dieser Zeit unterschiedlicher nicht hätte aussehen können. Trotzdem bist Du die Person an die ich mich wende, wenn ich verstanden werden möchte. Danke, dass Du bei all meinen Abenteuern stets an meiner Seite bist.
Liebe Linda, auch uns verbindet eine langjährige und treue Freundschaft. Seitdem wir gemeinsam ins Abenteuer ETH gestartet sind teilen wir gemeinsame Heiterkeit und ernste Gespräche, wenn wir mal wieder versuchen uns im Leben zu orientieren.
Danke für deinen aufmerksamen Beistand besonders in den letzten Wochen.
Bei meinen Freundinnen Stefanie, Helena, Petra, Bettina und allen weiteren Freunden möchte ich mich für gemeinsam erlebte kurze und lange Abenteuer bedanken, die mir immer eine willkommene Abwechslung im stürmischen PhD- Alltag waren.
Lieber Jens, lieber Martin, trotz der Entfernung zwischen uns seid ihr immer da für mich, wenn ich euch brauche. Ich bin euch unendlich dankbar für unsere Geschwister-«Liebe mit direkter Kritik», die in den letzten Jahren stetig gewachsen ist und die sich aus meinem Leben nicht wegdenken lässt. Wer sonst könnte mich immer wieder daran erinnern, dass das Leben mit mehr Leichtigkeit lustiger wird?
Euch, Mama und Papa, danke ich für den stabilen Rahmen, in dem ihr uns habt aufwachsen lassen. Es ist dieser stabile Hintergrund, aus dem heraus ich mein Leben lebe und auf den ich jederzeit zurückgreifen darf, wenn ich Sicherheit brauche. Danke dass ihr uns alle drei so kontinuierlich und verlässlich in unserem Lebensweg unterstützt.
Yvonne
v
vi
vii
Hydrogels comprise a class of soft materials that consist of a three-dimensional, crosslinked polymer network with large amounts of water tightly incorporated in their structure. The combination of polymer and high water content provides hydrogels with exceptional properties, such as diffusion of gases and molecules, transparency, strength and lubricity. With this unique combination of characteristics, the material exhibits similarities to human body tissue like no other existing synthetic material. Since the 1960s, hydrogels have found applications in the medical sector and are expanding into a constantly growing field. The diverse current and future applications range from spray bandages and contact lenses to intelligent drug-delivery systems, injectable tissue replacements, as well as implants in all parts of the human body.
Along with the ever-increasing field of application, however, the demands placed on the material are also growing. Despite the growing importance of hydrogels, fundamental research is still needed in the structural analysis of this class of materials. Detailed knowledge about the exact structure of a hydrogel and the significance (role) for its properties would enable (facilitate) the development of application-specific material designs in the future.
This thesis contributes to an in-depth structural elucidation of vinylic hydrogels by exploring, in detail, the relationship between the polymerization behavior of the monomers and the resulting polymer structure and macroscopic properties.
Monitoring the free radical polymerization reaction of polyacrylamide by UV/vis- absorption spectroscopy led to the detection of a side reaction of oxygen with the
Abstract
viii
initiator system TEMED/APS. Compression measurements indicated that this by- product may reduce the strength of the hydrogel.
X-ray scattering and light transmission measurements were used to characterize the cross-linking of the polymer network. The chain length and the mesh size between cross-linking knots were calculated by means of common polymer theories using the swelling behavior and the shear modulus of hydrogel samples. Results were compared with SEM images. A pronounced tendency of the crosslinker monomer to self-react and agglomerate produces a highly heterogeneous polymer distribution that is highly concentration dependent. The combination of all network components considered in this work thus leads to a picture involving a hierarchical organization of the hydrogel structure. This structure comprises crosslinker agglomerates with sizes ranging from the 10 nm range to the µm range, polymer chains connecting them and water pockets enclosed by the network.
At the surface of hydrogels, the nature of the polymeric structure and the lubricating water film are key aspects of hydrogel lubrication. A combination of neutron reflectometry and infrared spectroscopy were used to probe polymer volume fraction from the interface into the bulk hydrogel and its dependence on the molding material. The depth-dependence of the polymer-network density influences the compressibility of the hydrogel surfaces, as was demonstrated by both AFM- and micro- indentation. By changing molding materials, substantial differences in the gradient of polymer-network density were observed with depth. The lower the volume fraction of polymer at the hydrogel surface, the more water it can maintain at its interface as an incompressible film that is stable even under static conditions.
Such films render the hydrogel highly lubricious, with a speed-independent friction coefficient of µ = 0.01, measured in gemini contact. This result provides experimental evidence that the presence of these highly lubricious water films is strongly dependent on the polymer-network structure at the surface.
ix
Hydrogele sind weiche Materialien, die aus einem dreidimensionalen, vernetzten Polymernetzwerk bestehen, in dessen Struktur große Mengen Wasser eingebunden sind. Diese Verbindung von Polymer und hohem Wassergehalt verleiht Hydrogelen außergewöhnliche Eigenschaften wie zum Beispiel die Diffusion von Gasen und Molekülen, Transparenz, Stabilität und eine hohe Gleitfähigkeit an der Oberfläche.
Mit dieser einzigartigen Kombination an Eigenschaften weisen sie Ähnlichkeiten zu menschlichem Körpergewebe auf wie kein anderes existierendes synthetisches Material. Seit den 1960er Jahren finden Hydrogele deshalb Einsatz im medizinischen Bereich und erschliessen ein stetig wachsendes Aufgabenfeld. Die vielfältigen gegenwärtigen wie zukunftsträchtigen Anwendungen reichen von Sprühpflaster und Kontaktlinsen über intelligente Arzneimittelabgabesysteme bis zu einspritzbarem Gewebeersatz und Implantaten in allen Bereichen des menschlichen Körpers.
Zusammen mit einem immer grösser werdenden Anwendungsgebiet wachsen jedoch auch die Ansprüche, die an das Material gestellt werden. Trotz der stetig wachsenden medizinischen Bedeutung von Hydrogelen ist die Grundlagen- forschung noch immer mit der Strukturaufklärung dieser Materialklasse beschäftigt. Dabei würde exaktes Wissen über den genauen Aufbau der hydrogelen Struktur und deren jeweilige Bedeutung für die Eigenschaften des Hydrogels die Entwicklung anwendungsspezifischer Materialdesigns in der Zukunft erleichtern.
Die vorliegende Arbeit trägt zu dieser ausführlichen Strukturaufklärung bei, indem sie den Zusammenhang vom Polymerisationsverhalten der Monomere und ihrer Konzentration über die entstehende Polymer-Struktur bis hin zu makroskopischen
Zusammenfassung
x
Eigenschaften wie Transparenz, Festigkeit und Gleitfähigkeit des entstehenden Hydrogels detailliert erforscht.
Dabei führt die Beobachtung der freien radikalischen Polymerisationsreaktion von polyacrylamide, einem vinylen Hydrogel, mittels UV/vis- Absorptionsspektroskopie zur Identifikation einer Nebenreaktion von Sauerstoff mit dem Initiatorsystem TEMED/APS. Kompressionsmessungen lassen darauf schliessen, dass dieses Nebenprodukt die Festigkeit des Hydrogels schwächt.
Mittels Röntgenstreuung und Licht-Transmissionsmessungen wird die Vernetzung des Polymernetzwerks charakterisiert. Zusätzlich wird über das Schwellverhalten und das Schermodul von Hydrogelproben mittels gängiger Polymertheorien die Kettenlänge und die Maschengrösse zwischen den Vernetzungspunkten berechnet und mit SEM Aufnahmen abgeglichen. Eine ausgeprägte Tendenz des Vernetzer- Monomers zur Eigenreaktion und Agglomeration in Abhängigkeit der Konzentration führt zu einer stark heterogenen Verteilung des Polymers. Das Zusammenfügen aller in dieser Arbeit berücksichtigten Netzwerk-Komponenten führt dadurch zu dem Eindruck einer hierarchisch aufgebauten Gesamtstruktur des Hydrogels. Diese Struktur besteht aus Vernetzer-Agglomeraten mit Grössen vom 10-fachen nm Bereich bis in den µm Bereich, verbunden durch Polymerketten die zusammen mit dem Vernetzer Wasserporen umschliessen.
An der Oberfläche von Hydrogelen ist die Beschaffenheit der Polymerstruktur und der Wasser-basierte Schmierfilm ein Kernpunkt für die Gleitfähigkeit des Materials. Eine Kombination von Neutronenreflektometrie und Infrarotspektroskopie wird verwendet, um den polymeren Volumenanteil von der Grenzfläche bis ins Hydrogel-Innere zu erforschen und dessen Abhängigkeit von der Polymerisationsumgebung zu prüfen. Sowohl durch AFM- als auch durch Mikrokompressions-Tests wird demonstriert, wie die Veränderung der Polymernetzwerkdichte mit der Probentiefe die Kompressibilität der Hydrogel- Oberflächen beeinflusst. Ein Wechsel der Polymerisationsumgebung führt zu wesentlichen Unterschieden im Gradienten der Polymer-Netzwerkdichte mit der Tiefe.
xi
Je geringer der Volumenanteil des Polymers an der Hydrogel-Oberfläche ist, desto mehr Wasser kann es an seiner Grenzfläche halten. Solch ein inkompressibler Wasserfilm ist auch unter statischen Bedingungen stabil. Diese Filme machen das Hydrogel mit einem geschwindigkeitsunabhängigen Reibungskoeffizienten von
! = 0,01, gemessen im Geminikontakt, hochgradig gleitfähig. Dieses Ergebnis liefert den experimentellen Nachweis, dass das Vorhandensein dieser stark schmierenden Wasserfilme wesentlich von der Polymer-Netzwerkstruktur an der Oberfläche abhängt.
xii
xiii
1.1 Cartilage ... 1
1.2 Hydrogels... 4
1.3 Hydrogel Tribology ... 5
1.4 Scope of the Thesis ... 7
Bibliography ... 9
2.1. Introduction ... 11
2.2. Measurement Methods ... 12
2.2.1. Ultraviolet-visible Spectroscopy ... 12
2.2.2. Acoustic Resonance Spectroscopy ... 13
2.3. PAAm Hydrogel Polymerization Mechanism ... 14
2.3.1. Radical Formation in the TEMED/APS Initiation System... 14
2.3.2. Polymerization Process ... 16
2.4. Experimental Section ... 16
2.4.1. Free-Radical Polymerization of pAAm Hydrogel ... 16
2.4.2. TEMED Amine-oxide Synthesis ... 16
2.4.3. UV/vis Spectroscopy ... 17
2.4.4. Acoustic Resonance Spectroscopy ... 18
2.4.5. Indentation Measurements ... 18
Contents
Abstract ... vii1. Introduction... 1
2. PAAm Hydrogel Polymerization ...11
xiv
2.5. Results and Discussion ... 19
2.5.1. Following the Chemistry of FRP of pAAm Hydrogel in situ ... 19
2.5.2. Following the Polymer-Network Structure Formation of pAAm Hydrogel in situ ... 24
2.5.3. Effect of the Cross-linker Concentration on the Polymerization Process ...30
2.6. Conclusion ... 33
Bibliography ... 35
3.1. Introduction ... 37
3.2. Measurement Methods ... 38
3.2.1. X-ray Scattering ... 38
3.2.2. Visible Light Transmittance ... 40
3.3. Experimental Section ...41
3.3.1. Polymerization of pAAm Hydrogel ...41
3.3.2. X-ray Scattering ...41
3.3.3. Visible Light Transmittance ... 42
3.3.4. Scanning Electron Microscopy... 42
3.3.5. Swelling ... 43
3.3.6. Relaxation Measurements ... 43
3.4. Results and Discussion ... 45
3.4.1. Cross-linker Distribution in the Hydrogel Polymer Network ... 45
3.4.2. Polymer Chain-Length of the Hydrogel Polymer Network ... 57
3.4.3. The Structure of the Hydrogel Polymer Network ... 63
3.5. Conclusion... 68
Appendix 3.I. Calculations for the Polymer-Domain-Size Approximation ...70
Appendix 3.II. Calculations for the Chain Length and Mesh Size Approximation ... 73
Appendix 3.III. Measurement and Calculation Results ... 75
Bibliography ... 77
3. The Structure of the pAAm Polymer Network ...37
xv
4.1. Introduction ... 81 4.2. Measurement Methods ... 84 4.2.1. Neutron Reflectometry ... 84 4.2.2. ATR-FTIR Spectroscopy ... 85 4.3. Experimental Section ... 87 4.3.1. Mold Preparation ... 87 4.3.2. Polymerization of pAAm Hydrogel ... 87 4.3.3. Mold Characterization ... 87 4.3.4. Neutron Reflectometry ... 88 4.3.5. Infrared Spectroscopy ... 89 4.3.6. Nanoindentations ... 90 4.3.7. Microindentations ... 91 4.3.8. Friction in Gemini Contact...93 4.4. Results and Discussion ... 94 4.4.1. Substrate Properties before and after Polymerization ... 94 4.4.2. Polymer Volume Fraction at the pAAm Hydrogel Surface ... 95 4.4.3. Elastic Modulus of pAAm Hydrogel ...102 4.4.4. Friction of pAAm Hydrogel in Gemini Contact ... 105 4.5. Conclusion ... 107 Bibliography ... 109 4. The Structure of Hydrogel Surfaces ... 81
5. Conclusions ... 113 6. Outlook ... 117 Curriculum Vitae ... 121
1
Cartilage is a class of connective tissue in the human body. The structure of this outstanding natural material has evolved over millions of years into a specialized architecture that fulfils a multitude of specific functions. Its extracellular matrix mainly consists of proteoglycans and collagen fibres. It is avascular and aneural and the only cells found within cartilage tissue are chondrocytes. Depending on its characteristic structure and their associated functions, cartilage is classified as:
i) Elastic cartilage, which has a large amount of elastic fibres providing stable but flexible support to body parts such as the ear. It can undergo large elastic deformations but always returns back into its original shape.
ii) Fibrous cartilage, which is a matrix containing a large amount of collagen fibres. It is located between bones where it functions as a shock-absorbing cushion due to its high degree of fibre alignment. A prominent example for this cartilage type is the intervertebral disk.
iii) Articular cartilage (from Latin articulus = “small connecting part”), a special type of Hyaline cartilage, which covers the bone surfaces in synovial joints. It plays a major role in the lubrication of the joints.1 With a thickness of about 2 mm and a Young’s modulus between 20 – 30 kPa (Scheme 1.1),2-3 articular cartilage protects the joints from direct bone-to-bone contact, thanks to its tough and flexible nature. It supports the body load acting on the joint and, together with the surrounding synovial fluid, lubricates it to ensure
1. Introduction
1.1 Cartilage
2
the free movement of, for example, the knee, where typical relative speeds reach 100 mm s-1.4
From what is known about the structure of cartilage today, it is divided into four main zones (Scheme 1.2): a radial deep zone close to the bone surface with a fibrous alignment normal to the surface, a transitional middle zone, in which the fibre alignment changes towards a tangential orientation in the third, tangential superficial zone, and the articular surface on top. In addition, some works propose a superficial gel-like layer covering the articular surface.5
In comparison to industrial, man-made tribological systems, the lubrication of cartilage does not mainly depend on a velocity- and force-dependent fluid-film formation between contacting surfaces, but is driven largely by polymer-chain interactions surrounded by a water-based lubricant. As with every biolubricating system in nature, the interplay of articular cartilage and synovial fluid is based on three main principles. Firstly, sliding partners in nature generally have a compliant Scheme 1.1: The elasticity of soft tissues in the human body. Reprinted from Discher et al (2009).1
Articular surface
Tangential (superficial) zone
Transitional (middle) zone
Radial (deep) zone
Scheme 1.2: Schematic illustration of the structural gradient of bulk cartilage.
Reprinted from Crockett (2009).6
3
3
surface layer. Loading of such surfaces causes an elastic deformation that results in an increased contact area and thus reduces the pressure acting on the material under load. Secondly, the water-based lubricant, synovial fluid, is enriched with proteoglycan molecules. With their hierarchical bottlebrush structure comprised of proteins and polysaccharides (Scheme 1.3), they lead to an increase in viscosity of the synovial fluid. Glycoproteins help to overcome the poor lubricating character of water by trapping it at the cartilage surface. 5, 7 Third, the cross section of two biological bodies in sliding contact reveal a structural gradient towards their surfaces (Scheme 1.2).
Despite these generally acknowledged principles, the exact lubrication function of articular cartilage is not completely understood to date and remains the topic of ongoing research.4, 8-9 Three main theories are most commonly discussed for a systematic understanding of this complex structure-property relationship:10 One is the traditional engineering approach of fluid-film lubrication. Here, pressurization of the synovial fluid due to the relative velocity of surfaces in sliding contact leads to the formation of a hydrodynamic fluid film. Another theory is boundary lubrication. Here, the role of highly hydrophilic proteoglycans in synovial fluid and the existence of a superficial gel-like zone on the hydrogel surface are discussed as the main contributors to cartilage lubrication. A third theory describes cartilage lubrication by the pressurization of the interstitial fluid. Here, the main idea is that the interstitial fluid in a joint, and not the cartilage surfaces themselves, carry most Scheme 1.3: Bottlebrush structure of proteoglycan aggregate as it is present in synovial fluid. Reprinted from Lee et al. (2008).4
4
of the applied load under compression, both under a steady-state condition and in motion.
Hydrogels are three-dimensional, cross-linked hydrophilic polymer networks with a large amount of water tightly incorporated in their structures. Although the first mention of the word "hydrogels" appeared as early as 1894,11 they were created as the material as which we know them today only in the post-war era of the 1950s and 1960s, when the general materials class of polymers came into existence. One of the first applications of hydrogels was in gel electrophoresis,12 today a common screening method for biological macromolecules. Another early application, contact lenses,13 harks back to the early interest in employing polymers for permanent contact with living tissues. The requirements for contact lenses are as diverse as they are demanding: transparency paired with permeability of oxygen, high water content and lubrication, as well as flexibility, resistance to protein fouling, and non-inflammatory behavior. Evidently, hydrogels were targeted towards bio-based areas from the very beginning—a challenging field, in which the benefits of the polymeric network structure in combination with a high water content can be utilized to full advantage. Until now, no other synthetic material comes close to this unique combination of properties that can even mimic body tissues. It is the polymer network structure that is able to keep the water in the system and thus significantly defines their past and future applications in medicine.
The similarity between natural body tissue and synthetic hydrogels has played a major role in the development and research of these materials from the very beginning. Hydrogels not only expand the healing possibilities in the medical field, but by studying their structure-property relationships, it is also possible to draw conclusions about the structural components of biological tissues in relation to their functionalities. In this way, the fields of engineering and biology can benefit from each other, as this quote from the history of cartilage research demonstrates: “It seems probable that the physical laws which govern the behavior of lubrication in a
1.2 Hydrogels
5
bearing also apply to that in a joint, and reflection on our ignorance of the matter should lead to a healthy feeling of humility.” 14
In the decades following the development of hydrogels, much of the research has focused on elucidating the structure of the material. Today, the main interest appears to focus on developing the material further. To date, hydrogels have found their way into everyday uses such as care products, cosmetics or spray bandages, and are expanding the spectrum of possible applications in demanding medical areas such as intelligent drug-delivery systems, injectable tissue replacements, and implants in all parts of the human body. As a result, the industrial importance of hydrogels is growing steadily, as can be seen from the expansion in the range of hydrogel products of big companies in the implant market.15 Most recent hydrogel developments include a hydrogel-based, injectable bio-sensor that scans tissue chemistry for oxygen levels or detects viral infections.16 Towards the end of last year, an FDA-approved hydrogel was injected into the hearts of heart-attack patients, successfully restoring their heart function.17 At the beginning of this year, a research group from MIT reported on the successful development of a 3D- printable hydrogel-ink for fabricating brain electrodes.18 These inventions demonstrate the exciting possibilities these materials holds for the future.
While the evolution of hydrogels is progressing rapidly, it is sometimes astonishing to see how well the materials work, although the scientific community is still struggling with an in-depth insight into their microstructure. Fundamental research continues to further deepen our knowledge of the underlying principles of the multiplex structure of hydrogels.
Given the large interest in hydrogels in the biomedical and food industries, the tribological nature of hydrogels in combination with its mechanical properties play a major role in many of its applications. While the Stribeck curve for solid materials strongly depends on a fluid-film formation at the interface between two sliding partners with increasing speed and decreasing load, research into the tribology of soft sliding interfaces over the last twenty years presents a different picture. The
1.3 Hydrogel Tribology
6
tribology of gels and polymer brushes does not solely depend on a fluid film between contacting surfaces but seems to be largely driven by polymer-chain interactions.
Sliding hydrogels against different counter-surfaces led to a variety of hydrogel friction theories in literature. Gong suggested a gel friction mechanism called the
“repulsion-adsorption model” based on macrotribological investigations. In this model, friction is greater at slow sliding speeds when free polymer chains on the hydrogel surface have more time to adhere onto the counter-surface and deform elastically. For fast sliding speeds and repulsive surface interactions, a hydrated surface layer of the polymer network introduces a thin film of water between the sliding partners. According to her theory, this leads to a transformation from elastic friction to hydrated lubrication characterized by higher friction coefficients.19 Sawyer et al. proposed the concept of a transition load after investigating a hydrogel surface layer under AFM and in a microtribometer. Here, a load-independent friction coefficient is observed for small normal forces. This finding is in accordance with the study by Bielecki et al., in which they observed similar results for an oil-based polymer brush system 20. Based on the detection of stick-slip effects together with an increase in the friction coefficient above a particular normal load, Sawyer hypothesized a pressure-induced collapse of the polymer chains at the gel surface.21 In their later works, Sawyer et al. demonstrated the independence of the friction coefficient of two hydrogels in relative motion on the sliding speed up to a transition speed. The speed-independent low friction of so-called “Gemini interfaces” was studied with respect to polymer concentration and mesh size. From their observations, they predicted that thermal polymer-chain fluctuations on the hydrogel surface dominate friction processes at low sliding speeds and chain relaxations dictate speed-dependent, high friction between gel surfaces.22-25 Even though the above-mentioned theories account for the low friction between soft, polymeric materials, to date no precise explanation exists that could explain hydrogel friction in a universal/satisfactorily/all-encompassing manner.
7
The aim of this work was to elucidate the interrelation between the chemical polymerization processes with the structure of the polymer network and the mechanical and tribological performance of vinylic hydrogels. We believe that this approach is an essential step in the exact structural identification of the influence of individual polymer network components on particular hydrogel properties.
Decoupling the overall structure from the sum of its components can guide the improvement of individual properties without at the same time diminishing other properties related to performance.
In order to achieve this goal, polyacrylamide (pAAm) hydrogel produced via free- radical polymerization (FRP) was chosen as a study system in this work. Due to its established use in many applications, the choice of pAAm allowed us to focus on the underlying principles of structural influence on the performance of the hydrogel.
In the first part of the thesis, the polymerization process of pAAm hydrogel was studied in real-time. For this purpose, changes in the absorption behavior of the gelling solution were followed via UV/vis spectroscopy. This made it possible to monitor the conversion of monomer into polymer in a time-resolved manner.
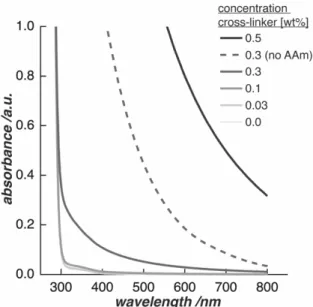
Differences were evaluated for varying hydrogel compositions. In this process, a by-product was identified when the reaction was performed in oxygenated environment. Its influence on the Young’s modulus of the hydrogel was investigated by means of microindentation analysis.
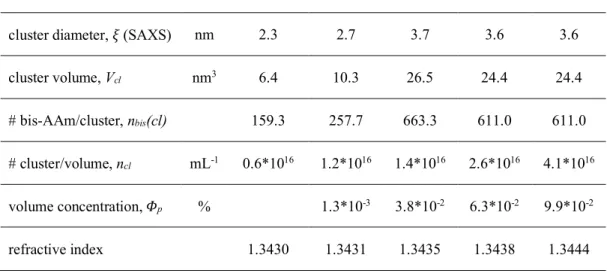
Closely associated with understanding the polymerization process was the goal of identifying the different hydrogel structures that polymerized as a function of the cross-linker concentration in the hydrogels. Here, the distribution of the cross-linker in the polymer network was analyzed on the nanoscopic and microscopic scales with small-angle X-ray scattering (SAXS) and light transmittance. These results were compared with calculations on the chain length and the mesh size of these networks. Extensive literature research complemented the obtained results, which together led to a revised picture of the polymeric network structure in free-radical polymerized vinylic hydrogels.
1.4 Scope of the Thesis
8
In the third and final part of this work, the focus shifts from the bulk hydrogel structure to the surface structure of aqueous gels and their polymeric state. By varying the polymerization environment, namely the molding conditions, the FRP of the hydrogels was altered, resulting in different surface characteristics. The relative amounts of polymer and water were analyzed down to a nanometer scale resolution by neutron reflectometry and infrared spectroscopy (IR). The results on this depth-dependent structural change were compared with results on the apparent modulus obtained from atomic force microscopy (AFM) and micro-indentation experiments. Ultimately, the dependence of friction coefficient on the surface structure was revealed in gemini contact.
We believe that this work will bring us closer to a solution of the long-standing challenge of unravelling the underlying polymer network structure that provides hydrogels with their unique properties.
9
1. Martini, F. H. T., M.J.; Tallitsch, R.B. , Human Anatomy 6th ed.; Pearson Education: San Francisco 2009.
2. Cohen, Z. A.; McCarthy, D. M.; Kwak, S. D.; Legrand, P.; Fogarasi, F.; Ciaccio, E. J.; Ateshian, G. A., Knee cartilage topography, thickness, and contact areas from MRI: in-vitro calibration and in-vivo measurements. Osteoarthritis and Cartilage 1999, 7 (1), 95-109.
3. Discher, D. E.; Mooney, D. J.; Zandstra, P. W., Growth Factors, Matrices, and Forces Combine and Control Stem Cells. Science 2009, 324 (5935), 1673-1677.
4. Burris, D. L.; Moore, A. C., Cartilage and Joint Lubrication: New Insights Into the Role of Hydrodynamics. Biotribology 2017, 12, 8-14.
5. Spencer, N. D., Aqueous Lubrication. World Scientific Publishing Company Singapore, 2014; Vol. 3.
6. Crockett, R., Boundary Lubrication in Natural Articular Joints. Tribol Lett 2009, 35 (2), 77-84.
7. Lee, S.; Spencer, N. D., Materials science - Sweet, hairy, soft, and slippery.
Science 2008, 319 (5863), 575-576.
8. McCutchen, C. W., Joints are not lubricated in the way Greene et al. say they are.
Proceedings of the National Academy of Sciences 2011, 108 (33), E461.
9. Jahn, S.; Seror, J.; Klein, J., Lubrication of Articular Cartilage. Annual Review of Biomedical Engineering 2016, 18 (1), 235-258.
10. Ateshian, G. A., The role of interstitial fluid pressurization in articular cartilage lubrication. J Biomech 2009, 42 (9), 1163-1176.
11. Van Bemmelen, J., Das hydrogel und das krystallinische hydrat des kupferoxyds.
Zeitschrift für anorganische Chemie 1894, 5 (1), 466-483.
12. Raymond, S.; Weintraub, L., Acrylamide gel as a supporting medium for zone electrophoresis. Science 1959, 130 (3377), 711-711.
13. Wichterle, O.; Lim, D., Hydrophilic gels for biological use. Nature 1960, 185 (4706), 117-118.
14. Shirley Jones, E., Joint Lubrication. The Lancet 1936, 227 (5879), 1043-1045.
15. https://investors.stryker.com/press-releases/news-details/2018/Stryker-acquires- HyperBranch-Medical-Technology-Inc/default.aspx (accessed 06/08/2020).
16. https://profusa.com (accessed 06/08/2020).
Bibliography
10
17. Traverse, J. H.; Henry, T. D.; Dib, N.; Patel, A. N.; Pepine, C.; Schaer, G. L.;
DeQuach, J. A.; Kinsey, A. M.; Chamberlin, P.; Christman, K. L., First-in-Man Study of a Cardiac Extracellular Matrix Hydrogel in Early and Late Myocardial Infarction Patients. JACC: Basic to Translational Science 2019, 4 (6), 659.
18. Yuk, H.; Lu, B.; Lin, S.; Qu, K.; Xu, J.; Luo, J.; Zhao, X., 3D printing of conducting polymers. Nat Commun 2020, 11 (1), 1604.
19. Gong, J. P., Friction and lubrication of hydrogels - its richness and complexity.
Soft Matter 2006, 2 (7), 544-552.
20. Bielecki, R. M.; Crobu, M.; Spencer, N. D., Polymer-Brush Lubrication in Oil:
Sliding Beyond the Stribeck Curve. Tribol Lett 2012, 49 (1), 263-272.
21. Dunn, A. C.; Uruena, J. M.; Huo, Y. C.; Perry, S. S.; Angelini, T. E.; Sawyer, W.
G., Lubricity of Surface Hydrogel Layers. Tribol Lett 2013, 49 (2), 371-378.
22. Dunn, A.; Sawyer, W. G.; Angelini, T., Gemini Interfaces in Aqueous Lubrication with Hydrogels. Tribol Lett 2014, 54 (1), 59-66.
23. Pitenis, A. A.; Uruena, J. M.; Schulze, K. D.; Nixon, R. M.; Dunn, A. C.; Krick, B. A.; Sawyer, W. G.; Angelini, T. E., Polymer fluctuation lubrication in hydrogel gemini interfaces. Soft Matter 2014, 10 (44), 8955-8962.
24. Urueña, J. M.; Pitenis, A. A.; Nixon, R. M.; Schulze, K. D.; Angelini, T. E.;
Gregory Sawyer, W., Mesh Size Control of Polymer Fluctuation Lubrication in Gemini Hydrogels. Biotribology 2015, 1–2, 24-29.
25. Dunn, A. C.; Pitenis, A. A.; Urueña, J. M.; Schulze, K. D.; Angelini, T. E.;
Sawyer, W. G., Kinetics of aqueous lubrication in the hydrophilic hydrogel Gemini interface. Proceedings of the Institution of Mechanical Engineers, Part H: Journal of Engineering in Medicine 2015, 229 (12), 889-894.
11
Free-radical polymerization is a straightforward approach commonly used in several research fields for the preparation of poly(acrylamide) (pAAm) hydrogels.
The advantages of the technique are numerous: the gel is easy to prepare, it polymerizes within minutes, and its preparation does not need elaborate laboratory equipment. PAAm hydrogels experienced a rise in popularity in the 1960s, when they were established as the base material for electrophoresis.1-4 The purpose of the hydrogel in this application is to sieve proteins and other biomolecules. In the same decade, hydrogels were introduced as a suitable material for contact lenses.5 In medical applications, a major advantage of hydrogels over conventional polymers is their oxygen permeability. These two advancements in the history of hydrogels are based on the material’s unique property of combining structural stiffness with the ability to transport both gases and molecules through a water-based matrix.
Interest in the research community was sparked and countless studies followed that investigated the microstructure of hydrogels and the influence of preparation parameters on structure.
Despite all efforts, no clear picture that has achieved recognition and validity across many disciplines has emerged of the detailed structure of these networks. However, it is generally acknowledged that the gel quality and its reproducibility strongly depend on preparation parameters such as temperature or the concentration of its components. While their ease of preparation and handling makes hydrogels convenient for use in many applications, this also carries the risk that the synthesis
2. PAAm Hydrogel Polymerization
2.1. Introduction
12
factors influencing the resulting network structure can be neglected. This motivated studies to improve sample quality and with it the reproducibility of experiments, by performing hydrogel preparation under oxygen exclusion.6 However, the influence of oxygen on hydrogel polymerization is seldom discussed in the literature.
The overall objective of this work is to shed light on the surface structure of hydrogels. In this chapter, we approach the topic by taking a closer look at the first step, namely the polymerization process itself. Here, we develop an understanding for a parameter that guides every hydrogel’s macroscopic property: the chemistry of the polymer network. In order to gain insight into this uncontrolled radical polymerization system, we follow the kinetics of the polymerization process on a chemical level with UV/vis spectroscopy and on a mechanical level with acoustic resonance spectroscopy. Knowledge of the polymerization mechanism of pAAm hydrogels will guide the preparation standard for this work.
UV/vis spectroscopy belongs to the class of energy-absorption spectroscopic methods. Its measurement range covers the entire wavelength spectrum of visible light (660 nm to 440 nm) and ultraviolet radiation (440 nm to <100 nm). During a measurement, the sample is scanned step-wise with light in this wavelength range.
When the light-beam hits an unbound or loosely bound electron in the chromophore of the sample molecule, the electron absorbs the introduced energy and is lifted from its ground state to an excited state. More strongly bound electrons need higher energy or smaller wavelengths for excitation than loosely bound electrons.
Consequently, electrons in double bonds are easier to excite than electrons in single covalent bonds. Hydrogel polymers have a low-energy absorption tendency in the UV/vis spectrum because their backbones are most often composed of carbon single bonds.
The UV/vis spectroscopy is a direct beam-measurement that records the intensities of the incident beam, I0, and the outgoing beam, I, after passing through the sample.
2.2. Measurement Methods
2.2.1. Ultraviolet-visible Spectroscopy
13
The difference between these two intensities (the attenuation) is then translated to the absorbance, A, or transmittance, T, of the sample with the Lambert-Beer law:
! = # ∗ % ∗ & (2.1)
! = log+,
-,
- = − log+,/ (2.2)
Here, # is the material specific extinction coefficient, c is the sample concentration and l is the sample thickness.
Acoustic resonance spectroscopy (ARS) is an impedance-based spectroscopic measurement method that detects mass displacements of a loudspeaker-hydrogel system upon sound excitation, related to the sample’s stiffness.7-8 It was recently developed in the group of Prof. Eric Dufresne at ETH. A loudspeaker, connected in series with a resistor and a lock-in amplifier, sends out a sound wave as the reference signal (Scheme 2.1.a). If a hydrogel is attached to the front of the speaker, the sound waves cause the hydrogel to vibrate. From this hydrogel oscillation, the speaker system experiences additional resonances. The hydrogel vibrates with varying intensity depending on its stiffness and the frequency of the sound input.
When the hydrogel vibrates to its first eigenmode, this special oscillation state corresponds to a mechanical movement of the hydrogel. The centre of mass of the hydrogel shifts and causes a force to be exerted on the speaker. The speaker- hydrogel resonance system is represented by a double spring-dashpot model
2.2.2. Acoustic Resonance Spectroscopy
Scheme 2.1: The electric circuit of the acoustic resonance spectroscopy consists of a speaker with the attached hydrogel sample and a resistor (a). Both speaker and hydrogel are modelled with a spring-dashpot element and a mass (b). Reproduced with permission from references 7,8.
14
(Scheme 2.1.b). The resulting speaker motion changes the current of the speaker coil and induces a voltage across it. The complex impedance of the system rises.
One can read out the eigenfrequency of a hydrogel-specific eigenmode from a frequency vs. impedance spectrum.
In combination with a finite-element model (FEM) of the first eigenmode of the hydrogel and a set of model data of hydrogel stiffnesses, the eigenfrequency is used to determine the Young’s modulus of the hydrogel:
0 = (2 ∗ 3 ∗ 4)6∗ 78 (2.3)
where E is the Young’s modulus, L is the gel width in its flask, 4 is the eigenfrequency of the first eigenmode and 7g is the density of the hydrogel.
If the measurement is repeated at regular time intervals during hydrogel polymerization, one can monitor the polymerization progress by reading out its stiffness-changes over time.
Ammonium persulfate (APS), generally denoted as the initiator of the reaction, dissolves in water freeing the ammonium (NH4+) and peroxide disulfate (S2O82-) ions. It is widely assumed today, that APS creates radicals through cleavage of the peroxide group in the molecule. In this case, the peroxide group splits to form two radicals from one APS molecule. However, this takes place only at elevated temperatures. At room temperature, the self-dissociation of APS is strongly reduced. By adding tetramethylethylenediamine (TEMED), generally denoted as the catalyst of the reaction, the radical formation is accelerated to a few minutes instead of hours or days at room temperature.
Already in the late eighties, it was proven that the addition of TEMED does not cause the APS to split into two peroxide radicals. Although often neglected, the work of Feng et al. proves that the addition of TEMED leads to the formation of a
2.3. PAAm Hydrogel Polymerization Mechanism
2.3.1. Radical Formation in the TEMED/APS Initiation System
15
free sulfate molecule and an intermediate product of a sulfate covalently bound to a TEMED molecule. The oxygen anion of the bound sulfate reacts with the hydrogen atom of a methyl group on the tertiary amine in TEMED. This change of bonding in the unstable molecule causes a cleavage between the sulfate oxygen and TEMED nitrogen. Two radicals are created: one from APS (Scheme 2.3, radical 1) and one from TEMED (radical 2). Together with a water radical (radical 3), they initiate the free radical polymerization of polyacrylamide.9-11
Scheme 2.2: A radical attacks the vinyl group of an AAm monomer. This process initiates free-radical polymerization, leading to the formation of a pAAm hydrogel.
Scheme 2.3: Radical formation from APS and TEMED. The molecules 1, 2 and 3 initiate the radical polymerization of pAAm.
TEMED APS
16
After their formation, the radicals attack the vinyl groups of the AAm and bis-AAm monomers in solution. This initiates chain growth and cross-linking of the pAAm hydrogel network (Scheme 2.2).
Stock solutions of the monomer acrylamide (AAm, Sigma-Aldrich, ≥99%), the cross-linker N,N’-methylenebis(acrylamide) (bis-AAm, Sigma-Aldrich, ≥99.5%), the initiation system tetramethylethylenediamine (TEMED, Sigma-Aldrich, 99%) and ammonium persulfate (APS, Sigma-Aldrich, ≥98%) were prepared in a deoxygenated environment (concentration O2 <100 ppm). The monomer, cross- linker and deoxygenated ultrapure water were mixed to final concentrations of 7.5 wt % AAm, and 0.0 / 0.03 / 0.1 / 0.3 / 0.5 / 0.8 wt % bis-AAm. The samples were named accordingly after their cross-linker concentration throughout this work, namely pAAm00, pAAm003, pAAm01, pAAm03, pAAm05 and pAAm08.
For the kinetic study, TEMED and APS (0.05 wt % respectively) were added to the monomer solution right before the start of the measurement under laboratory conditions (exposure to O2 environment, room temperature). Measurements were started immediately after solution transfer into a quartz cuvette (for UV/vis measurement) or a 25 mL vial (for acoustic resonance measurement) to follow the free-radical polymerization (FRP) from time zero.
Sample polymerization for comparative spectra was performed in a deoxygenated environment until full completion. In this work, we left hydrogels to polymerize for 24 h.
Hydrogen peroxide (11 mL, Merck, 30% solution) was added to 15 mL of ultrapure water and the mixture was heated to 60 °C under stirring. The solution was purged
2.3.2. Polymerization Process
2.4. Experimental Section
2.4.1. Free-Radical Polymerization of pAAm Hydrogel
2.4.2. TEMED Amine-oxide Synthesis
17
with compressed air, while a solution of TEMED (6.5 mL) and ultrapure water (5 mL) was added to the mixture dropwise. The reaction was carried out for four hours at 60 °C. It was cooled down to room temperature overnight. 1H-NMR spectra of the D2O-diluted reaction mixture confirmed the full conversion from TEMED to tetramethylethylene amine oxide (Figure 2.1).
The polymerization process was followed with UV/vis spectroscopy on two instruments (3 mL measurement liquid in a cuvette with a path length of 10 mm with Cary 60, Agilent Technologies, United States and 200 µL measurement liquid in a cuvette with a path length of 0.5 mm with V-670 Spectrophotometer, Jasco, Japan). The sample absorbance against the baseline of ultrapure water was recorded at room temperature over the entire wavelength range from 190 to 800 nm with a scan rate of 400 nm min-1. Spectra were recorded every two minutes for the first hour, every five minutes for the next 1.25 h, and every 30 min for the next 15 h of
2.4.3. UV/vis Spectroscopy
Figure 2.1:1H-NMR spectra of TEMED (a) and TEMED amine oxide (b) prove the conversion from TEMED to TEMED amine oxide with hydrogen peroxide. H2O* is residual H2O in D2O.
18
polymerization. For comparison, single spectra were recorded of samples completely polymerized in a deoxygenated environment.
The polymerization process was followed with acoustic resonance spectroscopy on an home-built instrument in the research group of Prof. Eric Dufresne.7 The acoustic resonance of a 10 mL hydrogel sample in a cylindrical, closed vial was scanned for frequencies of 10 – 300 Hz with a step size of 0.25 Hz. Spectra were recorded every two minutes for the first hour and once per hour for the next 15 h of polymerization.
PAAm hydrogel samples with cross-linker concentrations of 0.03 / 0.1 / 0.3 / 0.5 / 0.8 wt % and thicknesses greater than 1 cm were indented 24 h after polymerization and again after equilibrium swelling. Samples were covered in water during the measurements to prevent drying-out of the hydrogels. Four spots were indented on each sample.
Force-indentation curves were acquired to an indentation depth of 1 mm with an indentation speed of 0.01 mm s-1 (TA.XTplus Texture Analyzer, Stable Micro Systems, UK). A stainless-steel cylinder with a diameter of 2 mm was chosen as indenter to maintain the indentation area constant. The elastic modulus of every sample was derived by fitting the Hertzian model for a cylindrical indenter on an infinite half-space to each force-indentation curve.
9 = 2 ∗ 0 ∗ : ∗ ;
Here, F is the measured force, E is the elastic modulus of the sample, R is the cylinder radius, and d is the indentation depth.
2.4.4. Acoustic Resonance Spectroscopy
2.4.5. Indentation Measurements
19
The macroscopic properties of the final pAAm hydrogel are determined by two factors: First, the chemical composition of the polymer network and second, the polymer network structure. In order to gain a better understanding of both factors, we followed the free-radical polymerization process of pAAm hydrogels in situ. To this end, we monitored changes on a molecular level with UV/vis measurements during polymerization. We compared the results with the evolution of the mechanical strength of the forming network, as inferred from acoustic resonance spectroscopy (ARS). All results discussed in the following paragraph were drawn from pAAm samples with a cross-linker content of 0.1 wt%. This sample series is named pAAm01.
We followed the chemical reaction of the FRP of pAAm hydrogel in real time with UV/vis spectroscopy.
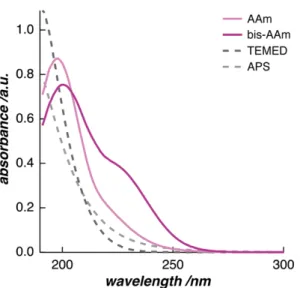
In a first step, we checked the detectability of every molecule in the reaction by the measurement technique. Measurements of diluted solutions of the monomer, AAm, and the cross-linker, bis-AAm, show the presence of the polymer components in the UV/vis energy spectrum (Figure 2.2). They both show a peak at the lower end of the UV/vis wavelength range (198 nm and 200 nm, respectively). Additionally, bis-AAm has a shoulder at 225 nm. AAm and bis-AAm monomers are detectable with UV/vis spectroscopy because they carry vinyl groups in their molecular structures. The free electrons of this carbon double bond absorb the incoming energy of the UV radiation.
In contrast, spectra of diluted solutions of TEMED and APS show that the initiation system of the FRP is transparent in the UV/vis region. This is due to the lack of
2.5. Results and Discussion
2.5.1. Following the Chemistry of FRP of pAAm Hydrogel in situ
Formation of pAAm Polymer
20
electrons in their molecular structures, that are free to absorb energy in the UV/vis- energy range. Here, the spectra only indicate an increasing tendency towards energy absorption just below 190 nm—the lower edge of the range of the measurement method.
When we recorded UV/vis spectra of the polymerization process in real time, the peak edges shifted towards lower wavelengths by approximately 20 nm over the first 30 min (Figure 2.3.a). This shift to shorter wavelengths over time corresponds to a reduction in peak size, i.e. to the consumption of monomer. During the polymerization, the vinyl group of AAm and bis-AAm monomers forms a covalent bond with another monomer in solution (Scheme 2.2). As the polymerization progresses, the concentration of energy-absorbing electrons in the vinyl groups decreases. This decrease causes a reduction in monomer-peak intensity. Since AAm monomers were the component with the highest concentration (7.5 wt %, 1.055 mol L-1) in the reaction solution, their consumption dominated the UV/vis reaction spectra. In order to confirm this conclusively, we measured aqueous monomer solutions with varying concentrations (Figure 2.3.b). Here, we demonstrated that the peak intensity reduced with a decrease in monomer concentration. As a consequence, the peak edges moved towards the peak center.
Figure 2.2: The UV/vis absorption spectra of the monomer, AAm, and the cross- linker, bis-AAm. The initiation system, TEMED and APS, appears to be mostly transparent to this measurement technique.
21
At high monomer concentrations, this phenomenon is observed as a shift of the right peak edge towards smaller wavelengths.
After 30 min, the shift indicating monomer consumption was accompanied by the formation of a shoulder at 287 nm. The shoulder reached its maximum absorption after 2.75 h polymerization time (Figure 2.4). This absorption shoulder demonstrates the formation of pAAm polymer. It evolved in parallel to the
Figure 2.4: After 30 min of polymerization, an absorption shoulder forms at 287 nm while the shift from monomer consumption in the upper UV/vis spectra continues.
Figure 2.3: The UV/vis absorption spectra of the first 30 min of the pAAm hydrogel polymerization are marked by a shift in the upper absorption range from 300 nm towards lower wavelengths (a). This shift displays a reduction in AAm monomer concentration causing both the peak position and the right edge of the peak to shift towards smaller wavelengths (b).
22
monomer consumption and is characteristic of pAAm in a UV/vis absorption spectrum.14 As with most polymers, pAAm contains only a small amount of chromophores. For this reason, the polymer shoulder appeared in the UV/vis spectra with a time delay; it was only after the monomer concentration had decreased and enough polymer had been formed that the 287 nm signal was revealed.
After the polymer shoulder reached its maximum absorption after 2.75 h polymerization time, we observed a reverse in the direction of the absorption shift in the upper absorption range. This seemingly opposite trend to the previously observed polymerization covered the polymer shoulder completely with advancing polymerization time. In addition, a peak at 340 nm evolved (Figure 2.5.a). We assume that these observations do not indicate a spontaneous decay of the newly formed polymer. If newly formed polymer decomposed back into monomers or oligomers, the radicals in the polymerization solution would have immediately initiated the formation of new polymer chains.
Thus, the reaction would have reached an equilibrium, reflected in the UV/vis spectra by a halt in absorption shift. Therefore, the backward shift in the UV/vis spectra is unlikely to be caused by an increasing concentration of free vinyl groups.
We also exclude the accumulation of carbonyl groups in the polymerizing solution as a cause for the rightward shift in absorption. The formation of C=O bonds in the system would produce a more dramatic change in the UV/vis spectra than the shift we observed.
Given these observations, it is highly plausible that a second chemical process takes place in the reaction solution, either in parallel to the actual polymerization reaction or with a time delay. All three findings of the second polymerization phase give an indication as to the process and product: First, the appearance of a peak in the UV/vis spectrum at 340 nm suggests the formation of a chemically new product.
Second, the shift in absorption towards higher wavelengths points towards the presence of free electrons in this new molecule.
Effect of Oxygen on the Synthesis Chemistry
23
All characteristics of this second synthesis were observed only for polymerizations in the presence of oxygen. Samples that were polymerized under a nitrogen atmosphere or in closed cuvettes did not show any of these two measurement characteristics (Figure 2.5.b). The only possible reactants in solution that are free to form a second product are the molecules APS, TEMED, oxygen, and water.
While APS and TEMED are important for the creation of radicals in the beginning of the first polymerization phase, they are not fully consumed during that step and remain in the reaction solution as hydrogen sulphate and TEMED (Scheme 2.2, 2.3).
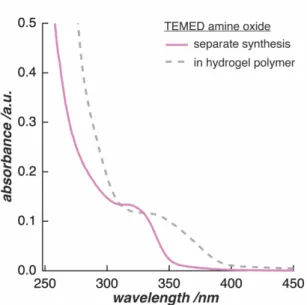
Subsequently, we hypothesize that the hydrogen sulphate reacts in the presence of oxygen to form persulfate and hydrogen peroxide.15-16 The accumulation of hydrogen peroxide in the reaction solution would then lead to the oxidation of the tertiary amine of TEMED.17 The reaction product is tetramethylethylenediamine dioxide, the amine oxide form of TEMED (Scheme 2.4).
This amine oxide meets all the criteria we have established from the measurement results: First, it is a new reaction product that absorbs UV light in the range of Figure 2.5: The UV/vis absorption spectra for long polymerization times of the pAAm hydrogel synthesis display a shift in absorption towards longer wavelengths and the formation of an absorption peak at a wavelength of 340 nm (a). If the polymerization is carried out in the absence of oxygen, the resulting UV/vis absorption spectrum shows the same pAAm-characteristic shoulder at 287 nm. It does not show a peak at 340 nm (b).
24
320 nm (Figure 2.6). The minor difference in absorbance between 320 nm and 340 nm is most likely caused by the difference in pH between pure water and the acidic polymerization environment. Second, TEMED amine oxide has free electrons in the molecule that can absorb UV light. We expect this absorption to cause the shift towards larger wavelengths in the higher absorption range.
From these observations, we conclude that the presence of oxygen is influencing the chemical processes in the free-radical polymerization of pAAm hydrogels.
However, its presence does not seem to influence the chemical composition of the polymer network itself. Instead, we hypothesize that the by-products of the additives TEMED and APS, namely hydrogen peroxide and TEMED amine oxide, are formed only when oxygen is present in the polymerizing solution.
Since they are only unbound by-products of the main polymer synthesis, they can be rinsed out of the network by immersing the hydrogel in water after completion of polymerization. In water, the hydrogel swells to equilibrium and all reaction residues are washed out of the polymer network. For this reason, we deduce that the influence of oxygen on the chemistry of the final pAAm hydrogel is negligible, if samples are properly rinsed with water after polymerization.
Scheme 2.4: Hydrogen sulphate reacts to hydrogen peroxide in the presence of oxygen. TEMED and hydrogen peroxide react to form amine oxide from TEMED.
TEMED amine oxide
25
2.5.2. Following the Polymer-Network Structure Formation of pAAm Hydrogel in situ
After revealing all chemical processes of the polymerization reaction of pAAm hydrogels, the question arises whether the presence of oxygen and TEMED amine oxide, while leaving the polymer chemistry untouched, may still have an influence on the polymer-network structure. In order to gain full understanding of the oxygen’s impact on the final hydrogel product after polymerization, we monitored changes in the hydrogel stiffness as the polymerization progressed.
In ARS measurements, we observed a continuous increase of the hydrogel stiffness for the first 30 min of polymerization (Figure 2.7.a). This measurement method records the vibrational response of the hydrogel to the incoming energy of a sound wave. From the ARS spectrum, the eigenfrequency can be determined for the first eigenmode that forms at the surface of the hydrogel sample upon excitation. For hydrogels that are physically cross-linked with entanglements but not chemically cross-linked, no eigenmode can be detected upon sound excitation with ARS. A
Formation of pAAm Polymer-Network
Figure 2.6: The UV/vis spectrum of separately synthesized TEMED amine oxide is characterized by a peak at 320 nm and leaping absorption below 300 nm. This spectrum shows the same characteristics as the final pAAm spectrum after polymerization completion, shifted only by 20 nm.