Research Collection
Doctoral Thesis
Small Molecule Activation by Dirhodium and Rhodium-Platinum Complexes
Author(s):
Jurt, Pascal Publication Date:
2021
Permanent Link:
https://doi.org/10.3929/ethz-b-000461532
Rights / License:
In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use.
ETH Library
Diss. ETH No. 27139
Small Molecule Activation by Dirhodium and Rhodium-
Platinum Complexes
A thesis submitted to attain the degree of DOCTOR OF SCIENCES of ETH ZURICH
(Dr. sc. ETH Zurich)
presented by Pascal Jurt
MSc ETH Chemistry, ETH Zurich Born on 16.10.1991
Citizen of Oberkirch LU, Switzerland
accepted on the recomandation of Prof. Dr. H. Grützmacher, examiner Co-Examiner: V. Mougel, co-examiner
2021
Timon and Pumbaa, in The Lion King
För mini Famelie, im Spezielle miis Mami.
I
A CKNOWLEDGMENTS
This work would not have been possible without the help and support of numerous people.
First of all, I would like to thank Prof. Hansjörg Grützmacher. Under your supervision, I was able to build on the research of master thesis in my PhD project. During my time at ETH, I learned a lot from you and was always deeply impressed by your enthusiastic and creative way of thinking about chemistry. Finally, I would like to thank you for sending me abroad, be it to conferences or to Russia for the PHIP measurements too.
I would like to thank Prof. Victor Mougel for agreeing to be the co-examiner of my PhD thesis. Also, you allowed me to use some of the special equipment of your laboratory.
Furthermore, I would like to thank Christine Rüegg, the heart and soul of the group.
You were always available to answer any dumb question we had. Thanks for your tremendous help with organizing trips and proofreading.
I would like to thank the two people who helped me a lot from the beginning of my thesis: Dr. Alexey Fedorov and Prof. Thomas Gianetti. They helped supervising me and especially, in the beginning, gave my work a clear direction. Alexey, without your help, my first paper would not have been published, and the connection to Novosibirsk would hot have been established. Thomas, you might have had the largest impact on my scientific career so far. Firstly, as a supervisor in my research project and my master thesis. Following, helping me in the first year of my PhD and still proof-reading everything I write, from paper drafts to important emails.
It is very important to me to highlight the work of my amazing students. My two master students, Anne Abels and Daniel Käch, did excellent work and are responsible for a significant amount of results presented herein. Anne was working on the Rh-Pt complexes and Daniel performed the luminescence study presented in this thesis.
Additionally, I would like to thank Patrick Müller, in the short time of your research project, you found some interesting and unprecedented results such as the aurodesilylation. Furthermore, you helped optimizing the reaction conditions for my tungsten-gold complex and investigating its properties.
Additionally, I would like to highlight the excellent work of Prof. Antonio Mezzetti. I liked the lab course already as a student and was from the beginning very proud to be teaching assistant in it. I learned a lot in the lab course, especially how to supervise other people. Additionally, you gave me the chance to work with you as your the organization of the lab course as well as a glimpse of ETH policy. Another positive side effect was that I learned how to engage with people in order to overcome their aversions in a mutually beneficial way. I believe all these experiences will enable me to progress further in my career.
Thanks to Dr. Hartmut Schönberg, you were always helpful with questions about safety. You always helped me to find a balance between not spending too much effort on unnecessary steps and being able to perform my reactions successfully and safely.
II Additionally, I would like to thank you for letting me use your picture in my public presentation, although you were not too pleased by the placement of a car next to it.
I would like to thank Dr. René Verel for the countless times you used your expertise to further my projects, which often contained a large number of stupid ideas for NMR measurements from my side. Thanks as well to the NMR assistants, that helped me with this: Dr. Matt Baker, Grégoire Le Corre, and Marcel Aebli.
Similarly, I would like to thank the scientists from the International Tomography Center in Novosibirsk. I am grateful for their generosity, allowing me to visit them and use one of their magnets for an entire week. The experiments I was able to do there led to parahydrogen induced polarization spectra that gave important insights into the reaction of my dirhodium complexes with hydrogen. I would especially like to thank Prof. Igor Koptyug, the head of the group, Kirill Kovtunov, senior scientist, and Nikita Chukanov, who helped with the preparation of the samples. I will always fondly thanks go to Oleg Salnikov, who was measuring the samples and fitted them afterward, so we could extract important coupling constants which would not have been accessible otherwise.
I believe I started to understand NMR during the years of my thesis, but X-Ray crystallography remains a mystery to me. I would like to thank the people who nevertheless helped me and my students with our structures for countless hours. A special thank here to Dr. Michael Wörle, Dr. Fabian Müller, Dr. Bruno Pribanic, and Frederik Eiler.
Similar to X-Ray crystallography, I remained ignorant about the beauty of DFT calculations. Thanks to the people who performed these for me: Dr. Jaap Borger and Juan José Gamboa-Carballo.
Thanks to the people who helped me with other analytical methods, Peter Kälin for EA, the MS service, Jonas Bösken, and Jan Bloch for cyclic voltammetry, Dr. Simon Rössler for GC-MS and Fabio Masero for Raman spectroscopy.
I would also like to thank all the people that worked with me in lab H122, who created a productive and enjoyable working environment. Thanks to Dr. Jieping Wang, Prof.
we are completely different, I always liked the atmosphere in the lab, and I had a lot of fun working with you.
A lot of people were involved with proof-reading this work, a very important task for which I am highly grateful. Thanks to Dr. Fabian Müller, Dr. Erik Schrader, Dr. Matt Baker, Clara Schweinzer, Raphael Bissig, Felix Graber, Nadine Jurt, and Frederik Eiler.
Besides very few exceptions, the Grützmacher group has been an amazing bunch of people these past few years. I will always remember our group hikes and sincerely apologize for those who barely survived in 2019. Thank you all for everything you have friends to me during the few years we were working together: Dr. Erik Schrader, Dr.
Fabian Müller, and Dr. Matt Baker.
III I would like to thank the friends from my earlier studies as we are still in contact. In our more or less frequent gatherings you allow me to see the scientific world from outside of the Grützmacher group. Thanks to Marcel Aebli, Simon Doswald, Emanuel Billeter, Stefan Kradolfer, Raphael Bissig, Christoph Neff, Andreas Gantenbein, Felix Graber, Daniel Hottinger, Roger Deplazes and Kristina Bodmer.
Special thanks to my friends at home. It was always enriching to see you on weekends non- and Kevin Kiser, although we usually all have very tight schedules, we managed to meet from time to time and we always had a very good time together.
Most importantly, I would like to thank my family, who always supported me. First of all, thanks to my pa. Although my studies took longer, you always supported me on my path. We always had everything we needed to thrive, from childhood to this day. I would like to thank my sister who made my life a lot easier, especially by organizing important parts of my private life. I also always deeply enjoyed our skiing trips and dinners. Thanks as well to my granny Elisabeth, the first person to see that I was capable of an academic career.
A very special thanks go to my mum, who unfortunately did not make it to this day. You were the person I wanted the most at my defense, already before I knew that you would not make it. I owe you so much. All through my childhood until I was a young adult you have always got my back. Because of you I never had to worry about anything and could just enjoy my life. Thank you.
IV
L IST OF A BBREVIATIONS
Å Angström
ACN Acetonitrile
BArF- Tetrakis[3,5-bis(trifluoromethyl)phenyl]borate
BB Broadband
BCDB B-(cyclodiborazanyl)amine borane BCP Bond critical point
BCTB B-(cyclotriborazanyl)amine borane COD 1,5 cyclooctadiene
COSY Correlation spectroscopy
CP Cross polarization
CTB Cyclotriborazane
DCM Dichloromethane
DFB 1,2-Difluorobenzene DFT Density functional theory
DME Dimethoxyethane
DSC Differential scanning calorimetry EDA Energy decomposition analysis
eq. Equivalent
EXSY Exchange Spectroscopy
GWP Global warming potential
GC-MS Gas chromatography-mass spectroscopy
GC-TCD Gas chromatography-thermal conductivity detector HMBC Heteronuclear multiple bond correlation
HMQC Heteronuclear multiple quantum correlation HOMO Highest occupied molecular orbitals
HSAB Hard and soft acids and bases
HSQC Heteronuclear single quantum correlation HTE High throughput experimentation
IR Infrared spectroscopy
IRC Intrinsic reaction coordinate
V
J Coupling constant
LUMO Lowest unoccupied molecular orbital MALDI Matrix assisted laser desorption MAS Magic angle spinning
MO-LCAO Molecular orbitals-linear combination of atomic orbitals NMR Nuclear Magnetic Resonance
NOCV Natural orbital for chemical valence NOESY Nuclear Overhauser spectroscopy NTf2 Trifluoromethanesulfoneimide OTf Trifluoromethylsulfonyl
PHIP parahydrogen induced polarization
Piv pivaloyl, C(=O)tBu.
ppm parts per million ppb parts per billion
QMID Quasi multiple ion detection
QTAIM quantum theory of atoms in molecules SEM Scanning electron microscopy
SOC Spin orbit coupling
TGA Thermogravimetric analysis
THF Tetrahydrofuran
TMS Trimethyl silyl
TOF Time of flight
Tp BH(pyrozol-1-yl)3
Trop 5H-dibenzo[a,d]cyclohepten-5-yl UV-VIS Ultraviolet-visible light
VT Variable temperature
XRD X-ray diffraction
VI
L IST OF C OMPOUNDS
VII
VIII
IX
P RESENTATIONS AND P UBLICATIONS
Parts of this thesis were published in the following Research article:
P. Jurt, O. G. Salnikov, T. L. Gianetti, N. V. Chukanov, M. G. Baker, G. Le Corre, J.
E. Borger, R. Verel, S. Gauthier, O. Fuhr, K. V. Kovtunov, A. Fedorov, D. Fenske, I.
V. Koptyug, H. Grützmacher -valent homobimetallic Rh complexes: influence of ligands on the structure and the intramolecular reactivity of Rh-
Chem. Sci. 2019, 10, 7937.
2019, 73.
The work in this thesis was presented at the following conferences and symposia:
SCS Fall meeting 2017, University of Bern, Switzerland, poster presentation:
23rd European Conference on Organometallic Chemistry, 2019, University of Helsinki, Finland, oral presentation:
mpounds for Metals on Carbon
12th International School of Organometallic Chemistry, 2019, University of Camerino, Italy, poster presentation:
metallic Rh-Rh or Rh-
SCS Fall meeting 2019, University of Zurich, Switzerland, oral presentation:
Rh-Rh and Rh-Pt Complexes
LAC Christmas symposium, ETH Zurich, Switzerland, oral presentation:
Dirhodium(I) and Rhodium(I)-
SSCI Meeting, 2020, ETH Zurich, Switzerland, poster presentation:
new Rhodium-
Public Presentation of the candidate, 2020, ETH Zurich, oral presentation:
2O to Benign Molecules
X
T ABLE OF C ONTENTS
1 GENERAL INTRODUCTION... 1
1.1 DINUCLEAR COMPLEXES AS POWERFUL HOMOGENEOUS CATALYSTS ... 1
1.2 ADVANCED NMRTECHNIQUES RELEVANT TO THIS THESIS ... 3
1.2.1 HMQC Experiments for Low Nuclei ... 3
1.2.2 Parahydrogen Induced Polarization ... 4
2 DIRHODIUM COMPLEXES ... 7
2.1 INTRODUCTION TO DIRHODIUM COMPLEXES ... 7
2.1.1 Dinuclear Rhodium(I) Complexes as Models for Heterogeneous Rh/C Catalysts ... 7
2.1.2 Dehydrogenation of Ammonia borane with Mono- and Dinuclear Rhodium(I) Complexes ... 8
2.1.3 Previous Work by the Group ... 10
2.2 RESULTS AND DISCUSSION ... 11
2.2.1 Synthesis of Bis(diphenylphosphino)methane Adduct 8 and Comparison of Analytical Data ... 11
2.2.2 Reactivity of Bis-triflate Complex 7 with H2 ... 14
2.2.3 Reactivity of dppm Adduct 8 with H2 ... 21
2.2.4 Semihydrogenation of Alkynes Catalyzed by Dirhodium Complexes ... 30
2.2.5 Determination of the Catalytically Active Species by 31P NMR Spectroscopy and Kinetic Competition Experiments ... 35
2.2.6 Dehydrogenation of Ammonia Borane with Dirhodium Complexes ... 40
2.3 CONCLUSION AND OUTLOOK ... 52
3 RHODIUM-PLATINUM COMPLEXES FOR THE REDUCTION OF NITROGEN OXIDES ... 53
3.1 INTRODUCTION ... 53
3.1.1 Environmental and Health Effects of N2O, NO and NO2 ... 53
3.1.2 Sources of N2O and NOx ... 54
3.1.3 Thermodynamic Basics for the Reduction of N2O, NO and NO2 with Hydrogen ... 54
3.1.4 Reactivity and Coordination Chemistry of N2O, NO and NO2 ... 55
3.1.5 Strategies for the Abatement of N2O and NOx... 58
3.1.6 Previous Work by the Group ... 60
3.2 RESULTS AND DISCUSSION ... 61
3.2.1 Synthesis of new Rh(I)-Pt(II) Complexes ... 61
3.2.2 NMR Spectroscopy of Compounds 16-24... 63
3.2.3 Single Crystal X-Ray Analysis of Compounds 16-24 ... 67
3.2.4 Theoretical Description of the Rh-Pt Bond ... 70
3.2.5 Catalytic Reduction of N2O and NOx with Hydrogen ... 73
3.2.6 Mechanistic Investigation ... 77
3.2.7 Attempted Synthesis of Dirhodium Complexes with Ligand 16 ... 87
3.3 CONCLUSION AND OUTLOOK ... 88
4 COINAGE METAL COMPOUNDS OF TMS/PHCC2TROPPPH2 ... 89
4.1 INTRODUCTION ... 89
4.1.1 Gold-Tungsten Complexes ... 89
4.1.2 Coinage Metal Complexes for Photoluminescence Applications ... 89
4.1.3 Previous Work of the Group ... 91
4.2 RESULTS AND DISCUSSION ... 91
4.2.1 Synthesis of Gold-Tungsten Complexes ... 91
XI
4.2.2 Abstraction of Chloride from 28... 93
4.2.3 Comparison of the Optical Properties of 28 and 31 ... 95
4.2.4 Abstraction of the Chloride from 27 ... 96
4.2.5 Cyclization of the PhC C Substituted Ligand 16 ... 98
4.2.6 Synthesis and Characterization of Coinage Metal Complexes 36 - 37 ... 100
4.2.7 Photoluminescence of the Coinage Metal Complexes 34, 36 and 37 ... 105
4.2.8 Cyclovoltammetry of 16, 34, 36 and 37 ... 109
4.3 CONCLUSION AND OUTLOOK ... 110
5 GENERAL CONCLUSION AND OUTLOOK ... 112
5.1 LIGAND DESIGN ... 112
5.2 SCOPE OF METALS USED IN THIS THESIS ... 113
5.3 ADVANTAGES OF SUBSTITUTED TROP LIGANDS FOR MONOMETALLIC COMPLEXES ... 113
6 EXPERIMENTAL ... 114
6.1 GENERAL REMARKS ... 114
6.2 COMPOUNDS OF CHAPTER 2 ... 117
6.2.1 Optimized Conditions for Compounds 1-7 for Completeness and Further Reference 117 6.2.2 Compounds Synthesized and Characterized as Part of this Thesis ... 125
6.3 COMPOUNDS OF CHAPTER 3 ... 131
6.4 COMPOUNDS OF CHAPTER 4 ... 153
6.5 HTEEXPERIMENTS ... 168
6.5.1 General Procedure 1 ... 168
6.5.2 General Procedure 2 ... 168
6.5.3 Results ... 169
6.5.4 Catalytic Hydrogenations in Acetonitrile ... 184
6.6 REDUCTION OF NITROGEN,NITRIC AND NITROUS OXIDE ... 185
6.7 CRYSTALLOGRAPHIC TABLES ... 186
7 REFERENCES ... 208
8 CURRICULUM VITAE ... 216
XII
A BSTRACT
Supported rhodium catalysts are known as one of the most active catalysts for hydrogenation and dehydrogenation reactions. However, the mode of interaction between Rh sites and H2 is still not fully understood. To gain further insight, a series of bimetallic complexes bearing new Rh(I) dimer moieties was developed, such as b and c (Scheme 1a). In comparison to heterogeneous catalysts, such model compounds allow for the characterization of the products by solution NMR techniques including parahydrogen induced polarization (PHIP) or 103Rh NMR spectroscopy.
Scheme 1. a) Synthesis of dirhodium complexes b and c, b) addition of hydrogen to complex b, c) transfer of hydrogen to the ligand of complex b.
Ligand a was designed to anchor two late transition metals in close proximity, enforcing a short intermetallic distance. This allows to form bimetallic complexes with dative intermetallic bonds such as dirhodium complexes b-f. With bis(diphenylphosphino)- methane (DPPM) as additional ligand, complex d adds hydrogen reversibly in solution.
Detailed DFT calculations allowed to gain insight into this hydrogen addition (Scheme 1b). The results are consistent with in situ NMR experiments. Remarkably, d activates H2 in a very similar manner as was calculated for small rhodium clusters. It is therefore a viable model compound for heterogeneous rhodium catalysts. Other complexes such as b transfer hydrogen to unsaturated C,C bonds of the ligand in a stepwise manner forming e and f (Scheme 1c). These reactions can show conceivable pathways of hydrogen transfer to a carbon support which may be relevant for Rh/C hydrogenation
XIII catalysts. The compounds were further investigated as hydrogenation catalysts for the semihydrogenation of alkynes and the dehydrogenation of ammonia borane.
Nitrous oxide (N2O) is a greenhouse gas 300 times more potent than carbon dioxide and an ozone depleting agent. Nitric oxide (NO), in combination with nitrogen oxide (NO2) is a major air pollutant and responsible for acid rain, ozone depletion and causes respiratory diseases. The abatement of such gases is therefore vital for industrial processes and car exhaust after-treatment systems. Nitrous oxide is commonly abated at high temperature and pressure over various catalysts, while nitric oxide and nitrogen oxide are reduced in modern exhaust after-treatment systems by ammonia over a titanium, zirconium or vanadium oxide catalysts. Homogeneous catalysts that can deplete these compounds are rare.
Scheme 2 a) Synthesis of heterobimetallic Rh-Pt complexes h and i, b) stepwise catalytic reduction of NO2 to N2.
A family of new Rh(I)-Pt(II) complexes of general structure h and i was synthesized.
The Rh(I)-Pt(II) core is chelated by ligand g. In this ligand phenyl substituents are bound to the alkynes, while ligand a bears trimethylsilyl groups. The phenyl groups were shown to enhance the stability of complexes h and i towards water and oxidizing conditions. In 195Pt NMR the bond between the two metals results in acoupling of 1JPtRh
= 83.4-109.3 Hz. A solvent molecule or a coordinating anion such as a chloride or a triflate completes the ligand sphere of the rhodium center. These complexes were shown to be active in the reduction of nitrogen dioxide, nitric oxide and nitrous oxide by hydrogen under mild conditions. Additionally, the mechanism for the formation of the N=N bond in the transformation from nitric oxide to nitrous oxide was shown to proceed through a spectroscopically characterized Rh(III)-Pt(II) hyponitrite complex.
XIV
Z USAMMENFASSUNG
Rhodium Nanopartikel auf Kohlenstoff zählen zu den aktivsten Katalysatoren für Hydrogenierungen. Es ist jedoch noch nicht vollständig geklärt wie das Wasserstoffmolekül mit der Rhodiumoberfläche interagiert. Da dies besser an molekularen Modellen untersucht werden kann, wurde eine Reihe von Dirhodium- Komplexen synthetisiert. Im Gegensatz zu heterogenen Katalysatoren können solche Modelle in Lösung mit NMR spektroskopischen Methoden wie Parawasserstoff induzierte Polarisation (PHIP) oder 103Rh NMR Spektroskopie untersucht werden.
Schema 1. a) Synthese der Dirhodium-Komplexe b und c, b) Addition von Wasserstoff durch Komplex c, c) Wasserstofftransfer auf den Liganden von Komplex b.
Der Ligand a wurde so entworfen, dass zwei elektronenreiche Übergangsmetalle sehr nahe bei einander koordiniert werden können. Dies führt zu einem kurzen intermetallischen Abstand. Durch Koordination von zwei Rhodium-Zentren pro Ligand a wurden die Komplexe b-f synthetisiert, welche sich durch eine dative Metall-Metall- Bindung auszeichnen. Mit Bis(diphenylphosphino)methan (DPPM) als zusätzlich verbrückender Ligand addiert Komplex c reversibel Wasserstoff (Schema 1).
Zusäztlich zu NMR Methoden wurde diese Addition mittels DFT-Berechnungen untersucht. Die Addition des Wasserstoffmoleküls durch den Dirhodium-Komplex d ähnelt der berechneten Wasserstoffaddition an kleine Rhodium-Cluster. Der Komplex d stellt somit ein gutes Modell für Rhodium-Oberflächen heterogener Katalysatoren
XV dar. Reagiert Komplex c mit Wasserstoff, wird dieser zu den ungesättigten C-C- Bindungen des Ligands transferiert. Zwei Moleküle Wasserstoff werden schrittweise auf den Liganden übertragen, wobei Komplexe e und f gebildet werden (Schema 1).
Diese Reaktionen zeigen auf, wie in heterogenen Katalysatoren Wasserstoff von Rhodium-Partikel zum Kohlenstoff-Trägermaterial transferiert werden kann. Die beschriebenen Rhodium-Komplexe wurden weiter als Katalysatoren für die Semihydrogenierung von Alkinen sowie für die Dehydrogenierung von Aminoboran eingesetzt.
Distickstoffmonoxid (N2O) ist ein 300-mal potenteres Treibhausgas als Kohlendioxid und trägt zum Ozonabbau in der Stratosphäre bei. Stickstoffmonoxid (NO) und Stickstoffdioxid (NO2) sind verantwortlich für sauren Regen, Ozonabbau und manche Erkrankungen der Atemwege. Der Abbau dieser Verbindungen ist somit von grosser Bedeutung wenn sie bei industriellen Prozessen als Abfallprodukt anfallen und in Abgassystemen bei Fahrzeugen. Distickstoffmonoxid wird normalerweise bei hoher Temperatur und Druck durch verschiedene Katalysatoren abgebaut. Die Stickoxide NO2 und NO werden in modernen Abgassystemen mit Ammoniak über Titan-, Zirkon- oder Vanadiumoxid-Katalysatoren reduziert. Nur wenige Komplexe sind bekannt, die diese Reaktion in homogener Lösung katalysieren können.
Schema 2. a) Synthese der heterobimetallischen Rh-Pt Komplexe h und i, b) schrittweise Reduktion von NO2 zu N2, katalysiert durch die Rh-Pt Komplexe.
Es wurde eine Familie von neuen Rh(I)-Pt(II) Komplexen mit der generellen Struktur von h und i, synthetisiert. Die Rh(I)-Pt(II)-Einheit wird vom chelatisierenden Liganden g stabilisiert. In diesem Liganden sind Phenylreste an die Alkine gebunden, während Ligand a Trimethylsilylgruppen aufwies. Die Phenylsubstituenden erhöhen die Stabilität der Komplexe h und i gegenüber Wasser und oxidierenden Bedingungen.
Die Rhodium-Platin-Bindung führt in der 195Pt NMR Spektroskopie zu einer Rhodium- Platin Kopplung von 1JPtRh = 83.4-109.3 Hz. Ein Lösungsmittelmolekül oder ein koordinierendes Anion vervollständigen die Koordinationssphäre des Rhodiums. Die
XVI synthetisierten Komplexe sind aktive Katalysatoren für die Reduktion von NO2, NO und N2O unter milden Bedingungen.
1
1 G ENERAL I NTRODUCTION
1.1 D
INUCLEARC
OMPLEXES ASP
OWERFULH
OMOGENEOUSC
ATALYSTSFigure 1. a) Rhodium(II) acetate and its derivates are examples of well-established dinuclear catalysts for the cyclopropanation of olefins by diazo compounds. b)-e) Recent examples of dinuclear complexes: b) catalyzes the cyclopropanation of olefins with dichloroalkanes or dichloroalkenes and zinc as a reducing agent, avoiding potentially explosive diazo precursors, c) catalyst for the Z-selective semihydrogenation of alkynes, d) is used for hydrogenation for CO2, e) a catalyst for N2 reduction.
Dinuclear [1]
Despite numerous reports, only few complexes are widely used for catalytic applications. The most prominent example is a rhodium(II) acetate dimer and its derivates. Such complexes have been exploited for carbene transfer reactions.[2]
These and other recent dinuclear complexes typically outperform monometallic alternatives in terms of activity and selectivity, functional group tolerance. Furthermore, they require only low catalyst loadings.[3] The interest in dinuclear catalysts has recently seen a renaissance.[4] This led to the development of new dinuclear complexes for catalytic transformations such as diazo-free cyclopropanation,[5] small molecule activation,[6] hydrogenation,[7] hydroformylation[8] and C-C coupling reactions[9] (structures of the catalysts are shown in Figure 1). Notably, heterobimetallic complexes serve as models for the transmetallation step for C-C coupling reactions.
Chen et al. could model the transmetallation in Sonogashira reactions by alkyne bridged Au-Cu complexes.[10] Most of these dinuclear species fall into two groups:
Heterobimetallic early-late transition metal complexes. This group typically relies on ligands that combine both soft and hard centers. They feature a highly polar dative interaction from the late to the early transition metal or main group metal (Figure 1c, d).[11]
Symmetric dinuclear complexes with a core consisting of two mid-to-late transition metals (Figure 1a, b).[12] They often rely on symmetric bridging ligands. This leads to an apolar bimetallic interaction.[13] Complexes of this
2 group have been of particular interest for the understanding of reactivity of small clusters.[14]
If the dinuclear complexes are not symmetric, one metal is often acting as an electron donor to the second metal (e.g. Figure 1a, d, e, see Figure 2). Such intermetallic interactions can help to stabilize bimetallic complexes and key reactive intermediates in catalytic cycles. Additionally, these complexes and/or reactive intermediates are further stabilized by bridging ligands (Figure 1b).[15] Catalytic reactions relying on dinuclear complexes as catalysts can proceed through new reaction pathways because of these stabilized intermediates. The catalytic cycles in Figure 2 exemplify these advantages of dinuclear complexes as homogeneous catalysts: A dinickel complex is presented, where a vinyl ligand is stabilized by bridging the two nickel centers. In the second example, hydrogen is heterolytically split over the Ag-Ru bond.
This leads to two separate Ag/Ru-H species that then transfer each a hydrogen center to the substrate in separated reaction steps.
Figure 2. Two examples of catalytic cycles taking advantage of dinuclear catalysts for organic transformations. a) A reductive cyclopropanation, in which a reactive vinyl ligand is stabilized by bridging the two Ni centers, b) heterolytic cleavage of hydrogen in the Z-selective hydrogenation of diphenylacetylene by a Ru-Ag complex.
3
1.2 A
DVANCEDNMR T
ECHNIQUESR
ELEVANT TO THIST
HESIS1.2.1 HMQC E
XPERIMENTS FORL
OWN
UCLEIIn NMR, the sensitivity of a spin ½ nuclei is determined by the product of the gyromagnetic ratio (the ratio of the magnetic moment and the angular momentum of a particle) and its natural abundance. Therefore, having either a very low gyromagnetic ration (e.g. 103Rh or 107/109Ag) or low natural abundance (e.g. 15N) results in a very low sensitivity in NMR experiments. As the signal to noise ratio in NMR spectroscopy increases with the square root of the number of scans, this leads to prohibitively long experiment times when measuring 1D spectra of such nuclei. Low natural abundance can be counteracted by preparing labelled compounds. However, to measure low nuclei, normally 2D experiments such as heteronuclear multi quantum coherence (HMQC, Figure 3) are employed. This drastically shortens the experiment times for such nuclei (e.g. a 103Rh spectrum can be obtained in 1-2 h compared to 3-4 weeks).
Figure 3. Pulse sequence for a standard HMQC experiment as used in this thesis. The narrow black boxes represent 90° pulses, the wide box represents a 180° pulse and is responsible for
1H/31P decoupling in the indirect dimension.
The standard pulse sequence for HMQC is depicted in Figure 3. A 90° pulse on the
1H/31P 90° channel creates magnetization, which evolves under the effect of the heteronuclear coupling 1J¹H/³¹P-X. The first 90° pulse on the X channel generates multi quantum coherence (a term consisting of a 1H/31P and X spin operator). The second 90° pulse on the X channel generates back single quantum coherence on 1H or 31P that can be detected. The 180° pulse on 1H/31P can be used to decouple the indirect dimension, while in the direct dimension a standard multi pulse decoupling during acquisition can be applied. Being a much simpler experiment compared to HSQC (heteronuclear single quantum coherence), it shows crucial advantages for the h as shorter time between excitation and detection.
Additionally, the exact calibration of the pulses on the X channel is less important, since fewer pulses are needed. However, it leads generally to broader lines compared to HSQC. Since normally only a small number of NMR signals with similar chemical shifts are expected in metal NMR spectra, this is considered a minor problem.[16]
4
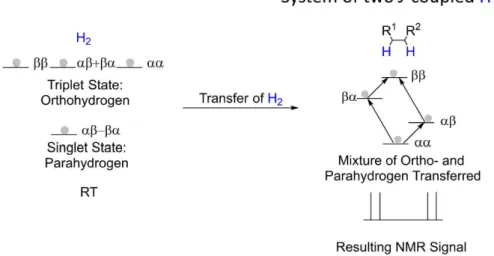
1.2.2 P
ARAHYDROGENI
NDUCEDP
OLARIZATIONalignment). For a set of two spins, the combined product-derived spin functions are . However, these functions need to be either symmetric or . However, simple linear combinations of them fulfill
- are symmetric with
respect to interchange and form a triplet nuclear spin state of hydrogen (called - is antisymmetric, and forms a singlet state (called parahydrogen). According to the Pauli principle, a wave function for two fermions (in this case hydrogen atoms) needs to be antisymmetric with respect to perturbation. It can be shown that therefore the symmetric spin states need to be multiplied with an asymmetric rotational wave function and vice versa. Therefore, the symmetric triplet state must be multiplied with the antisymmetric rotational wave function with rotational quantum number J=1 and the antisymmetric singlet state with the symmetric rotational wave function with J=0 (Figure 4).
This leads to two crucial consequences for parahydrogen induced polarization (PHIP):
The energy difference between the singlet state and the triplet state is around 10 times larger than in normal NMR experiments and does not require an applied magnetic field, since the energy difference arises from the different rotational wave functions. This allows reaching the low temperature limit, where the ground state is populated predominantly. For example, at 40K at thermal equilibrium, around 90 % of all hydrogen molecules are in the singlet state.
As the spin state and the rotational state must change simultaneously, the transition is spin forbidden and requires the presence of a paramagnetic catalyst to occur. A potential catalyst is for example iron oxide hydroxide (rust). If no paramagnetic compound is present, the parahydrogen can be warmed to room temperature and subsequently stored. It will not relax back to its thermal equilibrium state.
Figure 4. Simplified graphic showing the spin states of hydrogen and their equilibrium population at different temperatures. J denotes the wave number of the rotational wave function.
5 If normal hydrogen is transferred to a substrate and NMR spectrum is measured, the resulting signal is observed as two classical doublets coupled to each other. As all states are almost equally populated in hydrogen, this leads to an almost equal population of the four states of the two coupled 1H nuclei in the substrates. Additionally, all transitions are absorbing energy and are therefore observed as positive signals in an NMR experiment (Figure 5).
Figure 5. A normal NMR experiment with two J-coupled protons leads to two doublets in the NMR spectrum.
If parahydrogen is transferred to a substrate, only two states ( ) are populated, leading to a large signal enhancement. Additionally, two transitions absorb energy like in a standard NMR experiment (green arrows in Figure 6), but the two other transitions emit energy (red arrows in Figure 6). This leads to two antiphase doublets in the NMR spectra (Figure 6).[17]
Figure 6. A situation analogue to Figure 5, but parahydrogen is used instead of normal hydrogen consisting of 75 % orthohydrogen and 25 % parahydrogen.
6
1.2.2.1 E
XPERIMENTALS
ETUPAs soon as the parahydrogen is transferred to a substrate, the transition between the spin states is no longer spin forbidden and the molecule starts to relax to thermal equilibrium. The NMR experiment should therefore be started immediately after the substrate was in contact with parahydrogen. To ensure this, parahydrogen is normally bubbled through a solution inside the NMR magnet. Immediately after the parahydrogen addition is stopped, a single scan 1H NMR experiment with a 90° pulse is performed. A typical experimental setup is represented in Figure 7.
Figure 7. Experimental setup for PHIP experiments.[18]
7
2 D IRHODIUM C OMPLEXES
2.1 I
NTRODUCTION TOD
IRHODIUMC
OMPLEXES2.1.1 D
INUCLEARR
HODIUM(I) C
OMPLEXES ASM
ODELS FORH
ETEROGENEOUSRh/C C
ATALYSTSScheme 3. a) Hydrogen addition as calculated by DFT for small rhodium clusters on carbon support, b) dinuclear Rh hydrogenation catalyst (left) and its proposed hydrogen addition, according to DFT (right), c) a rare dirhodium(II) dihydride characterized by NMR spectroscopy.
Rhodium nanoparticles on support materials are widely used in heterogeneous catalysis and various industrial processes, especially for hydrogenation and dehydrogenation reactions.[19] Still, the mode of interaction between the supported rhodium sites and H2 is not fully understood.[20] A recent DFT study reported that hydrogen addition by small rhodium clusters on the surface of a carbon support features one bridging and one terminal hydride ligand (Scheme 3a). The resulting hydride can be transferred to the carbon support in an additional step.[21] However, such intermediates have not been observed experimentally to date. Low valent dinuclear rhodium complexes can serve as molecular models for supported Rh sites.
They help to understand the hydrogen addition mechanism on these materials.[11]
While the representation of supported heterogeneous catalysts by a dinuclear model significantly reduces their complexity, this approach allows for a reliable identification of reaction products. Despite the apparent oversimplification of the intrinsic complexity of heterogeneous catalysts, it is therefore insightful to study examples of well-defined dinuclear Rh(I)-Rh(I) complexes capable of activating H2, but such examples are scarce.[22] Scheme 3b presents one of these rare dirhodium(I) complexes. According
8 to DFT calculations, this complex activates dihydrogen, leading to one bridging and one terminal hydride ligand. The resulting hydrides resemble the hydrides on the supported rhodium clusters mentioned above. This mode of hydrogen addition dissymmetrizes the dirhodium complex while simultaneously forming a stabilizing Rh Rh bond.[23] Again, experimental evidence for such a dirhodium dihydride species is still lacking. However, non-symmetric complexes with a dirhodium(I) core containing chloride ligands in place of the hydrides were reported.[24] Additionally, a symmetric dirhodium dihydride complex, which forms an intermetallic bond, was characterized by NMR and IR spectroscopy (Scheme 3, c).[25]
In contrast to the exploitation of complexes with symmetric ligands, the advantages of non-symmetric ligands to control the reactivity of two adjacent Rh centers are underexplored.[26] A non-symmetric electronic environment imposed by the ligand could enable otherwise inaccessible reactivity manifolds.[11, 27] Further, modelling the environment and complexity of heterogeneous catalysts becomes possible with these non-symmetric ligands. For example, modelling of metal-support interfaces and surface defects. In particular, a non-innocent ligand with multiple unsaturated C-C bonds could provide insights into the reactivity of Rh/C interfacial sites of metallic Rh nanoclusters or nanoparticles on carbon-based supports.
2.1.2 D
EHYDROGENATION OFA
MMONIA BORANE WITHM
ONO-
ANDD
INUCLEARR
HODIUM(I) C
OMPLEXESThe dehydrogenation of ammonia borane has been intensively investigated in recent years due to the possible use of ammonia borane as hydrogen storage material (19.6 wt% of hydrogen).[28] To avoid the harsh conditions of thermal decomposition,[29]
homogeneous metal catalyzed dehydrogenation with Sc,[30] Y,[31] Ti,[32] Cr[33], Mo,[33-34]
W,[33] Re,[35] Fe,[36] Ru,[37] Os,[38] Co,[39] Rh,[40] Ir,[34b, 37a, 41] Ni,[42] Pd,[43] Th and U[44]
complexes have been investigated. Also metal-free catalysts have been reported.[45]
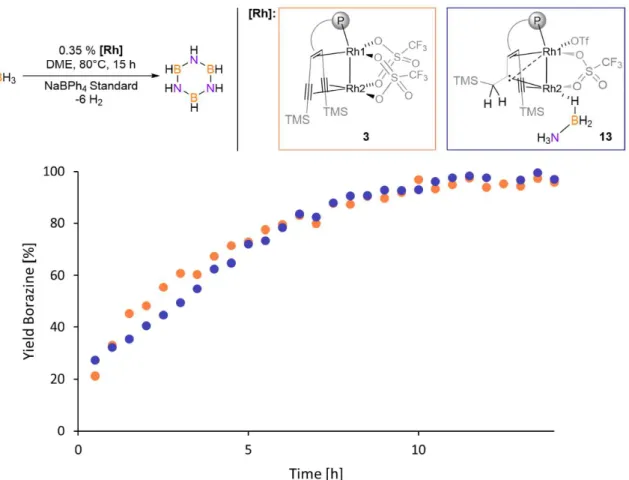
Up to 2.7 equivalents of hydrogen can be released by the most efficient catalyst at 1 % loading (Scheme 4).[37g, 46]
9 Scheme 4. The dirhodium catalyst discussed in this thesis in relation to the state-of-the-art catalysts and a diruthenium catalyst, investigated by F. Müller.[47]
Most catalysts that dehydrogenate unsubstituted ammonia borane form polyaminoborane, (NH2BH2)n,[34b, 36a, 36c, 37b, 37e, 38, 41] or a mixture of borazine and polycondensated borazine moieties.[36a, 37f-i, 42-43, 45a] The selectivity of this transformation depends on whether aminoborane, H2B=NH2, as first highly reactive intermediate formed in the first dehydrogenation step, remains coordinated to the metal
center or not -
growth mechanism. Upon release into solution, H2B=NH2 gives di-, tri- or tetramers which upon further dehydrogenation react further to borazine and polycondensed borazines.[48] Very few catalysts are able to convert H3N-BH3 selectively to borazine.[35a, 37h, 37i, 40a]
allow the formation of borazine in good to excellent observed yields (75 %[35a] and 99
%[37h, 37i] respectively, see Scheme 1) but relatively high catalyst loadings (1 and 5 mol%) were employed and the product was not isolated. Only once the isolation of borazine was reported and in this reaction. 1 mol% of [Rh2Cl2(COD)] (COD = 1,5- cyclooctadiene) was added but although the reaction proceeds with high selectivity, the isolated yield was only around 10 %. Additionally, the reaction was found to be catalyzed by rhodium nanoparticles.[40a] To date, the best borazine syntheses involve reacting sodium borohydride with ammonium sulfate in the presence of aluminum chloride as catalyst or the thermal decomposition of ammonia borane[49]. Borazine is formed in 67 % yield in both reactions.
Borazine and well-defined boron nitride (BN) nanosheets are potentially highly interesting precursors to generate thin layers of h-BN.[50] These layers find applications in, for example, UV light emitters,[51] or as components of polymer nanocomposite membranes for hydrogen separation[52] or as precursors to BCN (boron-carbon- nitrogen) ceramic materials.[53]
10
2.1.3 P
REVIOUSW
ORK BY THEG
ROUPScheme 5. a) TropPPh2 complexes for group 9 and 10, b) previously reported synthesis of ligand 5 and dirhodium complexes 6 and 7. [a] NaBH4, MeOH, 98 %. [b] TMSC CH, 5 mol%
[Pd(PPh3)4], CuI, toluene, Et3N, 76 %. [c] SOCl2, DCM, 0°C, 80 %. [d] LiPPh2, toluene, 63 %.
[e] [Rh2(µ-Cl)2(C2H4)2], benzene, 82 %. [f] AgOTf, DCM, 83 %.
Previous work by our group showed that the bidentate concavely shaped tropPPh2
-donor (Ph2 accepting binding site (C=Ctrop) enables strong binding[54] to several transition metal centers including Pd,[55] Rh[56] and Ir[56b, 57] (Scheme 5a). Additionally, a synthesis for an alkyne substituted trop-ligand 5 from known compound 1[58] and dirhodium complexes 6 and 7 were reported by Sébastien Gauthier, Florian Läng and Florian Puschmann.[59]
Ligand 5 was synthesized using a Sonogashira coupling in the key step. Then the benzylic alcohol is converted to a chloride and subsequently to a phosphine group (Scheme 5). Two Rh(I) centers were then coordinated to each ligand 5 by addition of [Rh2(µ-Cl)2(C2H4)2] leading to the bridged dimeric complex 6. Subsequently the chloride bridges were replaced by triflate anions, forming complex 7.
11
2.2 R
ESULTS ANDD
ISCUSSION2.2.1 S
YNTHESIS OFB
IS(
DIPHENYLPHOSPHINO)
METHANEA
DDUCT8
ANDC
OMPARISONOF
A
NALYTICALD
ATAScheme 6. Synthesis of bis(diphenylphosphino)methane adduct 8.
The syntheses of ligand 5 and dirhodium complexes 6 and 7 were optimized, which allowed to synthesize them on a multigram scale. Dirhodium complex 8 was formed in good yield in a ligand exchange reaction from 7 and bis(diphenylphosphino)methane (dppm) in dichloromethane. The structures of 6-8 were determined by X-ray diffraction methods and are presented in Figure 8.
12 Figure 8. Molecular structures of 6 (top left), 7 (top right) and 8 (bottom). For selected bond lengths and angles see Table 1. Non-coordinating triflate anions, hydrogen atoms and solvent molecules are omitted for clarity.
13 Table 1. Selected bond lengths (Å) and angles (°) for complexes6-8, ct: center of coordinated multiple bond.
bond length (Å) or angle (°) 6 7 8
All complexes possess a distorted square planar geometry around Rh1 and a nearly ideal square planar environment around Rh2 ( Rh1= 0.29, 0.46 and 0.37, and Rh2=0.06, 0.01 and 0.03 for 6, 7 and 8 respectively).[60] The Rh Rh distance in monomeric 7 is 2.6297(2) Å, which is 0.21 Å shorter than in dimeric 6 (2.8464(3) Å, Table 1). A similar bond length of 2.6880(4) Å was observed for the symmetric dirhodium compound [Rh2Cl2(tfepma)2(CNAr)], one of the rare examples of metal-metal bonded dirhodium(I) complexes (tfepma = bis(trifluoroethoxy)phosphinyl)methylamine, CNAr = p-F- C6H4NC).[61] This shortening of the Rh Rh bond is accompanied by an elongation of the P Rh bond from 2.1829(3) Å in the chloride bridged dimer 6 to 2.2115(5) Å in the monomeric triflate complex 7, indicating that the Rh Rh interaction in 7 is stronger. In contrast, the bond lengths of the coordinated C-C multiple bonds as well as the respective C-Rh distances are virtually identical in 6 and 7 (Table 1). Dppm adduct 8 has a Rh Rh bond length of 2.7691(7) Å, an intermediate value between those of 6 and 7. Additional significant differences are observed in the alkene and alkyne bonds trans to P2 and P3, which are elongated in 8 (Rh1-ct(C5-C6) 1.981(7) Å and Rh2- ct(C1-C2) 2.232(7) Å in 8 compared to Rh1-ct(C5-C6) 1.913(2) Å and Rh2-ct(C1-C2) 2.062(2) Å in 7). This can be explained by the stronger trans influence of the phosphine ligand.[62]
The 13C NMR chemical shifts of the olefins for the chloride bridged dimer 6 and the monomeric triflate complex 7 13C = 37.9 vs 37.0 ppm for 6 and 7, respectively). A strong shielding is observed for both triple bonds in the triflate complex, suggesting that Rh2 site in 7 is more electron-rich than in 6 13C = 99.4 to 86.6 ppm for TMS- C 13C = 74.0 to 65.3 for TMS-C 6 and 7, respectively). The Rh Rh bond can be best described as a dative bond where electron donation from Rh2 into the antibonding orbital of the Rh1-P bond occurs. This is similar to the bonding in early-late bimetallic transition metal complexes.[11] For complex 8, this theory is
14 supported by calculations, as the HOMO-2 and HOMO-3 orbitals show a clear overlap between the two metal centers, with larger orbital contribution of Rh2 (Figure 9).
Figure 9. Calculated orbitals for complex 8 in acetonitrile, which are relevant for the description of the intermetallic bond. These orbitals show significant orbital contributions in between the two metals. The larger contribution of Rh2 to the orbital indicates a dative bond from Rh2 to Rh1. Applied Functional: -D/def2-SVP.
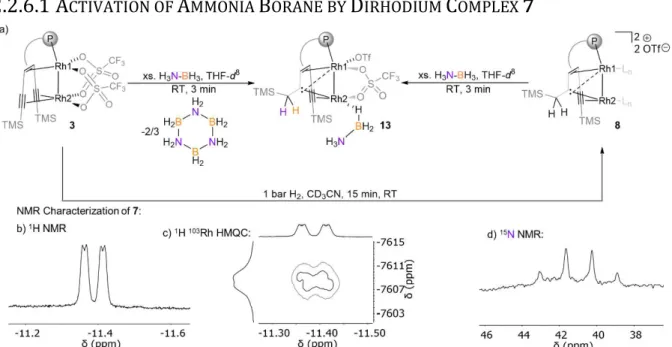
2.2.2 R
EACTIVITY OFB
IS-
TRIFLATEC
OMPLEX7
WITHH
2Scheme 7. Stepwise semihydrogenation of 7 in CD3CN at room temperature followed by in situ NMR spectroscopy. Ln is CD3CN.
In a J. Young NMR tube containing monomeric triflate complex 7 dissolved in CD3CN, 1-2 bar hydrogen was added to the headspace. Following the reaction progress by
15 NMR spectroscopy allows observing a stepwise hydrogenation of the two triple bonds of the supporting }2tropPPh2 ligand. The first alkyne semihydrogenation step proceeds quantitatively within 15 min and, strikingly, yields carbene species 9 (Scheme 7). This chemoselectivty in alkyne hydrogenation is uncommon and was only observed once for alkynes bound to d8 metal centers with a Pt(II)-( -H)3-Pt(II) core.[63]
The semihydrogenation of the remaining triple bond leads to a coordinated alkene 10 (Scheme 7). It proceeds with a slower rate, requiring ca. 20 h at 2 bar of H2 for quantitative conversion. The carbene complex 9 and the carbene alkene complex 10 display characteristic signals in 13C NMR spectra assigned to the bridging carbene at
13C = 171.1 and 166.3 ppm and the methylene carbon at 13C = 35.5 and 36.7 ppm, respectively. The newly formed methylene group is identified by the two diastereotopic proton signals in the 1H NMR spectrum at 1H = 2.38, 3.18 and 1.99, 3.03 ppm for 9 and 10, respectively, showing a geminal 2JHH coupling of 12.1 and 13.1 Hz. HMBC spectrum supports the structural assignment of complexes 9 and 10; this data is presented in Figure 10 and Figure 11.
Figure 10. 1H-13C HMBC spectrum of 9 in CD3CN. Cross peaks relevant for the assignment of 9: a) the TMS region, b) coupling of the methylene group to the carbene and C=C-CCarbene, and c) C=C signals coupling to the aromatic protons.
16 Figure 11. 1H-13C HMBC spectrum of 10 in CD3CN. Cross peaks relevant for the assignment of 10: a) signals coupled with the TMS region, b) signals coupling to the newly formed alkene -C=C carbon atoms, c) coupling of the methylene group with carbene and C=C-CCarbene carbon atoms.
The bridging nature of the carbene ligand shows two rather differentJCRh coupling constants for 9 (1JCRh = 33.9, 11.1 Hz) and for 10 (1JCRh = 35.7, 11.2 Hz), which suggests that the Rh2 center has a closer contact to the carbene carbon atom than Rh1 (Scheme 7). Results of single crystal X-ray analysis of 10 are consistent with the proposed structure, although the quality of data is rather poor (Figure 12). The observed 1JCRh coupling constants of 33.9 and 35.7 Hz are similar to the earlier reported values for dinuclear rhodium(I) complexes with bridging carbenes which likewise show 1JCRh in the range of 30-32 Hz.[4] The assignment of cis- semihydrogenation in 10 is supported by two 1H NMR signals at 1H = 2.58 and 5.07 ppm coupled to each other with 3JHH = 11.3 Hz.
17 Figure 12. Crystal structure of 10, triflate anions, hydrogen atoms and solvent molecules are omitted for clarity. The structure is of poor quality, bond lengths and angles are therefore not discussed.
The two olefinic carbon atoms of the central trop double bond in 6-8 (C5 and C6 in Figure 8) are strongly shielded, most likely due to the anisotropic effects of the neighboring alkyne group (the chemical shifts range from 13C = 37.0 - 48.7 ppm).
Upon hydrogenation, these carbon atoms show a remarkable difference in the chemical shift (9: 13C = 43.3 and 98.8 ppm, 10: 1 3C = 62.9 and 100.0 ppm for C6 and C5, see Scheme 7$). The difference in chemical shift in C6 is consistent with hydrogenation of the alkyne to an alkene, lowering its anisotropic effect. However, the strong deshielding of C5 in both complexes indicates an overlap between the C2 carbene p orbital and the trop double bond, leading to an allyl-like bonding around Rh1.[64] This inference is further confirmed by the JCRh coupling constants, which are much smaller for the C5 carbon atom than for the C6 carbon atom (Table 2, numbering in the trop ligand is according to Scheme 7). A similar bonding motif was reported with a dinickel(I) core in the solid state.[65] To summarize, complexes 9 and 10 contain a carbene carbon atom in conjugation with the central double bond of the trop ligand and this allyl-type ensemble is bridging to the Rh(I)-Rh(I) fragment. The coordination sphere of Rh2 in 9 and 10 in solution is likely stabilized by CD3CN.
18 Table 2. Selected NMR data of complexes 7,9, and10. Pos.: Position, see Scheme 7.
a) no proton showed a significant coupling to Rh2 in the 1H-103Rh HMQC spectrum Furthermore, a characteristic shielding is observed in the 31P NMR spectra when comparing the trop phosphorus signal at 31P = 104.6 ppm in the monomeric triflate complex 7 with the respective signals at 31P = 59.7 and 67.0 ppm in the carbene complex 9 and the carbene alkene complex 10. This change is accompanied by a shift to a smaller 1 coupling constant in 9 and 10 (1 = 127.2 and 136.0 Hz) as compared to that in 7 (1 = 185.6 Hz), indicating a higher trans influence of the Rh2 center in 9 and 10 compared to 7. This suggests weakening of the P Rh bond that is offset by strengthening of the Rh Rh bond, attributed to the interaction with the bridging carbene ligand.[66] The latter could also be viewed as a distorted dirhodacyclopropane.[67] Analysis of the 103Rh NMR data reveals that Rh1 is significantly shifted to lower frequency when comparing 7 with 9 and 10 ( 103Rh =
6852, 7184, and ppm for 7, 9 and 10, respectively), further supporting a more electron rich Rh core. Comparing 9 and 10 using the 103Rh NMR shift of Rh2 suggests that the Rh2 site in 10 is more electron rich than in 9 ( 103
9 and 10, respectively),[68] which is consistent with a change in the ligand sphere from
19 an alkyne to a weaker -accepting alkene. These results indicate that the dinuclear core becomes more electron rich with each hydrogenation step.
The semihydrogenation of the ligand was performed in a stepwise manner, where the carbene complex 9 was first formed under H2, followed by deuteration to the carbene alkene complex 10 under a D2 atmosphere. In this case, deuterium is only incorporated to the double bond (blue hydrogen atoms in Scheme 7) indicating irreversible ligand hydrogenation. This observation is consistent with the results of an experiment where 10 was formed in situ under an atmosphere of H2 and subsequently placed under an atmosphere of D2. In this case no deuterium incorporation is observed within 14 h, indicating that both hydrogenation steps are irreversible (Figure 13-Figure 14).
Figure 13. Top: 2H NMR spectrum of 10 formed from 9 (20.0 mg, 18.5 mol) in 0.5 mL acetonitrile in a J. Young NMR tube pressurized with 3 bar D2 after 24 h, a) D2, b) signals corresponding to the formed alkene C-D bonds, c) residual water (left) and solvent (acetonitrile, right). No deuterium label was detected on the methylene group. Bottom: 1H NMR spectrum of 10 for comparison.
20 Figure 14. Top: 2H NMR spectrum after 10 was exposed to 2 bar D2 for 2 h in 0.5 mL acetonitrile in a J. Young NMR tube, a) D2, b) signal could occur from minor H/D scrambling or unknown impurity, c) solvent (acetonitrile), d) water. Bottom: 1H NMR spectrum of 10. No deuterium label on the olefinic positions was detected.
21
2.2.3 R
EACTIVITY OF DPPMA
DDUCT8
WITHH
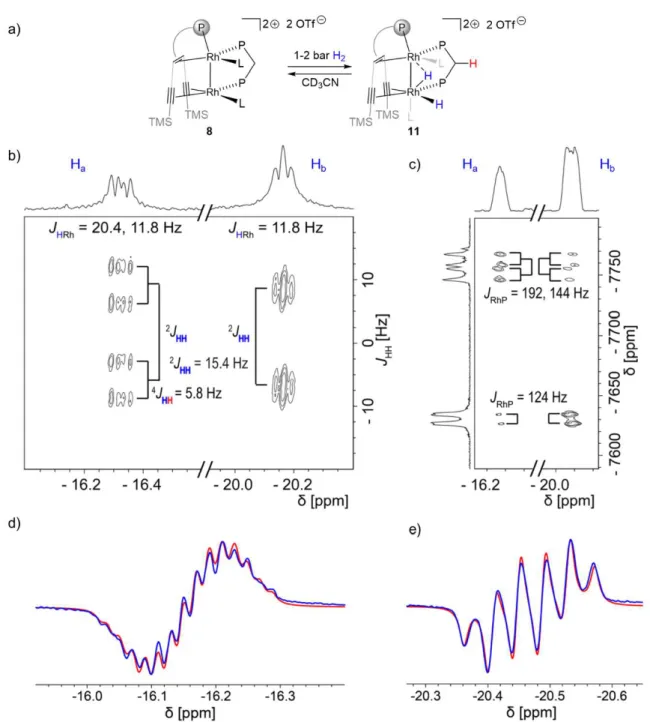
2Figure 15. Proposed addition of hydrogen by complex 8 (L is acetonitrile) and relevant spectroscopic data: a) reaction of 8 with hydrogen, as proposed on the basis of NMR experiments, b) J-resolved 1H{31P} spectrum, c) 1H-103Rh HMQC spectrum, d) PHIP 1H spectrum (blue) with simulation (red), of hydride Ha, e) PHIP 1H{31P} spectrum (blue) with simulation (red) of hydride Hb. The representation of ligand 5 was simplified in structures 8 and 11.
Next, 1-2 bar of H2 was added to a solution of dppm adduct 8 in CD3CN in a J. Young NMR tube. In this experiment hydrogen was not transferred to the ligand and no bridging carbene was observed. Instead, a mixture of 8 and a Rh dihydride complex (Figure 15a) is formed that is stable for several days at room temperature. Two characteristic hydride signals are observed at approximately 1H 16.2 and 20.5 ppm. Dihydride 11 was further characterized by low temperature NMR experiments as
22 well as using the PHIP technique. The removal of the H2 (or the D2) atmosphere from a J. Young NMR tube containing a mixture of the dppm adduct 8 and the dihydride 11 cleanly forms back 8, indicating a completely reversible hydrogen addition (Figure 16, Figure 17).
Figure 16. 1H NMR spectra (from top to bottom): 8 in CD3CN, after addition of 4.6 bar H2, after three freeze pump thaw cycles, under argon, after addition of 4.7 bar D2, after three freeze pump thaw cycles, under argon; a) solvent residual signal, b) inert solvents: dichloromethane and hexane, c) H2, d) TMS region (see Figure 17 for details), e) characteristic signals of 11, f) characteristic hydride signals.
23 Figure 17. Magnification of the TMS region of Figure 16, a) characteristic signals of 11, b) an unidentified side product.
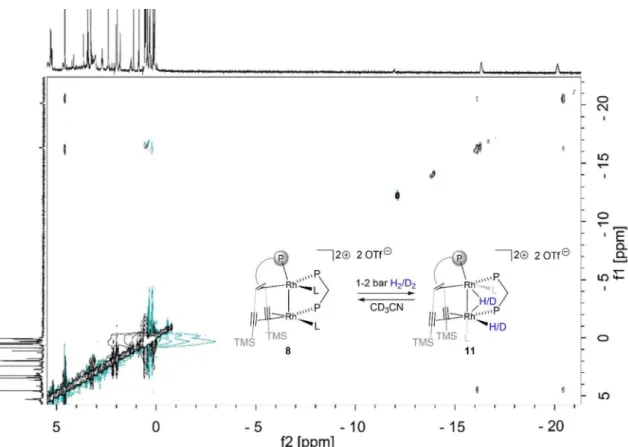
This reversible hydrogen addition of 8 is further supported by EXSY spectroscopy, variable temperature NMR spectroscopy and a partially negative line-shape (PNL) of the orthohydrogen peak in the PHIP NMR spectrum (Figure 18-Figure 20)[69] Since PNL does also occur in an identical PHIP experiments with 7, the same mode of hydrogen addition is likely occurring with both species, 7 and 8. Upon formation of cis- dihydride 11 103Rh1 = 7758 ppm is significantly shifted to lower frequencies. This low-frequency shift exceeds even the ones observed for 9
103Rh1 = 10 ( 103Rh1 = . The Rh2 nucleus ( 103Rh = 7630 ppm) shows likewise a strong shift to lower frequencies relative to the 103Rh2 nuclei in 9 and 10 103Rh2 = 6733 and 7066 ppm, respectively), which is in -donating hydrides on both metal centers (Figure 15b). 1H{31P} J-resolved 2D NMR spectroscopy revealed the JHH and JHRh
coupling constants (Figure 15c).
24 Figure 18. EXSY spectrum of in situ formed 11 under hydrogen atmosphere at 238 K shows cross peaks between the hydrides and hydrogen due to the reversible hydrogen addition.
Figure 19. Variable temperature 1H NMR spectra of the in situ formed 11 under hydrogen atmosphere measured at 25, 15, 5, 5, 15, 35 °C.
25 Figure 20. 1H PHIP NMR spectrum acquired after bubbling parahydrogen at 1 atm through a CD3CN solution of 7 and dppm, resulting in the formation of 8, (top) and the corresponding thermal spectrum after relaxation of hyperpolarization (bottom). Observed PHIP signals (2.38 ppm (JHH = 12.0 Hz), 3.17 ppm (JHH = 11.9 Hz)) are assigned to species 11due to similar chemical shifts and couplings to those of 9. A small orthohydrogen signal at 4.52 ppm with a PNL indicates reversible H2 addition.[69b] Two hydride signals of species 11 were observed at -20.47 ppm and -16.15 ppm.
The two hydrides remain coupled in complex 11, as revealed by the 2JHH = 15.4 Hz splitting in the indirect dimension. The hydride at 1H = 16.2 ppm shows an additional
4JHH = 5.8 Hz coupling to another proton, assigned by COSY to one of the methylene protons of the dppm ligand (red in Figure 2a). NMR characterization of 11 was completed by 1H, 13C, 19F, 29Si and 31P spectra. Altogether, this data confirmed that dppm adduct 8 adds hydrogen reversibly and is in equilibrium with the dihydride species 11. No semihydrogenation of the triple bonds is observed in this case.
Recording the J-resolved spectrum with 31P decoupling allows extracting the JHRh
coupling constants (Figure 15c). While the hydride at 1H = 16.3 ppm appears as a doublet of doublets (JHRh = 20.4, 11.8 Hz), the hydride at 1H = 20.2 ppm appears as a pseudo-triplet (JHRh = 11.8 Hz). The observed 2JHH coupling of 15.4 Hz is larger than typically found in traditional cis hydrides formed via oxidative addition (2JHH = 7.2-9.5 Hz),[70] suggesting another geometry since higher coupling constants indicate larger angles between the substituents. Overall, these results are consistent with a bridging geometry for the dihydride 11 (Figure 15a). Assignment of Ha cis to the intermetallic bond is based on the observation of a remarkable long-range 4JHH coupling of Ha to a CH2 proton (4JHH = 5.8 Hz, highlighted red in Figure 15a, b). Unfortunately, recording
26 a J-resolved spectrum with 103Rh decoupling was not successful due to the large difference in the chemical shift, and it was not possible to eliminate the JHRh couplings from both metal centers at once. Therefore, the JHP coupling constant could not be accessed with this approach. However, the 1H-31P coupling constants could be extracted by fitting the observed PHIP signals (Figure 15d, e). The PHIP spectrum in Figure 2e was recorded with a 31P decoupling, which selectively eliminates only 31P couplings arising from the dppm moiety. This allows distinguishing coupling constants between the hydrides and dppm/trop phosphorus centers. In addition, PHIP experiments allowed to determine the sign of the 2JHH coupling constant between the two hydrides as negative, 2JHH = 15.4 Hz.
The proton Ha shows a 2JHP coupling to the dppm 31P center of 2JHP = 13 and 6 Hz, indicating a cis 2JHP coupling.[70] The second hydrogen atom Hb is likely close to the cis position of the trop phosphine center, as suggested by the coupling constants of 2JHP
= 24 and 18 Hz. The coupling to the second dppm phosphorus center is substantially smaller (JHP = 4 Hz). However, the large JHH coupling constant as well as the similar ones to both Rh centers (resulting in the pseudo-triplet in Figure 15b) are consistent with an interaction with the second Rh center. This assignment allows reporting a cis coupling constant of 2JHRh = 11.8 Hz. This indicates that the close Rh Rh contact is preserved after the addition of dihydrogen. Having assigned Ha as the terminal hydride, while Hb interacts with the two Rh centers forming an unsymmetric hydrogen bridge, the Rh Rh core can be described either as a Rh(II)
polarized bond, due to the bridging nature of Hb. Both descriptions imply a close Rh Rh contact. It was tested whether similar hydrides can also be observed with other ligands. For this, tricyclohexylphosphine, diphenylphosphine oxide and triazabicyclodecene were added to dirhodium complex 7 subjected to conditions of the in situ PHIP experiments. In none of these experiments similar hydrides were observed. This demonstrates that only the dppm adduct 8 leads to observable rhodium hydrides upon hydrogen addition. However, all PHIP experiments with added ligands except triazabicyclodecene showed the hyperpolarized signals of methylene CH2
protons with very similar chemical shifts and coupling constants as observed for 9. In addition, a partially negative line shape signal for orthohydrogen was observed with all ligands, suggesting that hydrogen addition is reversible.
27
Scheme 8. DFT calculations (Gaussian09,B97X-D/def2-SVP) for the hydrogen addition pathways from8-MeCN to 8a-HHoxand 11-HHox. L is acetonitrile. The transition states were confirmed to connect the two respective energy minima by IRC calculations.