Zeitschrift für
Kristallographie
International Journal for Structural, Physical, and Chemical Aspects of Crystalline Materials
Volume 197 No. 1/2 1991
General Aspects W. Schwieger, K.-H. Bergk, D. Heidemann, G. Lagaly, Κ. Beneke Y. Ito, S. Kuehner, S. Ghose
A. J. C. Wilson
M. Rieder, Ζ. Weiss
Hochauflösende "Si-Festkörper-NMR-Unter- 1 suchungen am synthetischen Natriumsilicathydrat
Makatit und dessen kristalliner Kieselsäure
Phase transitions in leucite determined by high tern- 75 perature, single crystal X-ray diffraction
Space groups rare for molecular organic 85 structures: The arithmetic crystal class mmmP
Oblique-texture X-ray photographs: more informa- 107 tion from powder diffraction
Inorganic Crystal Structures W. A. Dolíase,
C. R. Ross II
Crystal structure of orientationally disordered Na2(Ca,Sr)Si04
H. Effenberger, H. Fuess, Crystal structure and hydrogen bonding in Li/H- G. Müller, T. Vogt
L. S. Dent Glasser, R. P. Gunawardane, R. A. Howie
K. H. Lii, C. H. Li, T. M. Chen, S. L. Wang
exchanged petalite, HAISi„O10
The crystal structure of sodium strontium silicate, Na4SrSi3Oe
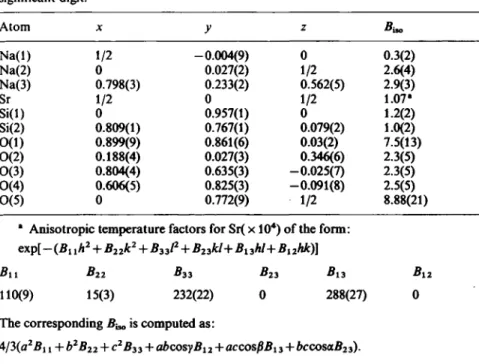
Synthesis and structural characterization of sodium vanadyl (IV) orthophosphate NaV0P04
13
27
59
67
(Continuation see cover p. 4)
R. Oldenbourg Verlag · München
Z. Kristallogr. 197,1 - 1 7 4 (1991) Indexed in Current Contents ISSN 0044-2968
Zeitschrift für Kristallographie
International Journal for Structural, Physical, and Chemical Aspects of Crystalline Materials
Editors-in-Chief:
Editorial Board:
H. Schulz, H. G. von Schnering.
With the assistance of W. Hönle
G. Bergerhofî, E. F. Bertaut, M. L. Fornasini, H. Fuess, M.Hart, P.Hartman, S.Haussühl, E.Hellner, R.J.Hill, K.Huml, Jiang Min-hua, F.Liebau, E.Parthé,
P.Paufler, S.Rundqvist, L.A.Shuvalov, Y.Takéuchi
Please send manuscripts Professor
Dr. G. BergerhofT Professor Dr.
Dr. h.c. E. F. Bertaut Professor
Dr. M.L. Fornasini Professor
Dr. H. Fuess Professor Dr. M. Hart FRS Professor Dr. P. Hartman Professor Dr. S. Haussühl Professor Dr. E. Hellner Dr. R. J. Hill Dr. W. Hönle Professor Dr. K. Huml Professor Jiang Min-hua Professor Dr. F. Liebau Professor
Dr. Dr. h.c. E. Parthé Professor
Dr. P. Paufler Professor St. Rundqvist Professor Dr. Dr. h.c.
H. G. von Schnering Professor
Dr. H. Schulz Professor
Dr. L. A. Shuvalov Professor
Dr. Y. Takéuchi
to one of the following addresses:
Institut für Anorganische Chemie, Universität Bonn Gerhard-Domagk-Straße 1, D-5300 Bonn 1, F.R.G.
Laboratoire de Cristallographie, CNRS Grenoble, 25, Avenue
des Martyrs, Β.P. 166X, Centre de Tri, F-38042 Grenoble Cedex. France Università di Genova, Istituto di Chimica Fisica
Corso Europa 26,1-16132 Genova. Italy
Fachbereich Materialwissenschaften, Fachgebiet Strukturforschung, Technische Hochschule Darmstadt, Petersenstr. 20,
D-6100 Darmstadt. F.R.G.
Department of Physics, Schuster Laboratory The University, Manchester M l 3 9PL. U.K.
Instituut voor Aardwetenschappen
Budapestlaan 4, Postbus 80.021, NL-3508 TA Utrecht. Netherlands.
Institut für Kristallographie, Universität Köln Zülpicher Straße 49, D-5000 Köln 1. F.R.G.
FB Geowissenschaften
Lahnberge, D-3550 Marburg/Lahn. F.R.G.
CSIRO Division of Mineral Products,
P.O. Box 124, Port Melbourne, Victoria 3207. Australia Max-Planck-Institut für Festkörperforschung
Heisenbergstraße 1, D-7000 Stuttgart 80. F.R.G.
Institute of Macromolecular Chemistry, Czechoslovak Academy of Sciences, CSSR-16206 Prague 6. Czechoslovakia.
Institute of Crystal Materials, Shandong University Jinan Shandong, PRC
Mineralogisch-Petrographisches Institut Olshausenstraße 40 — 60, D-2300 Kiel. F.R.G.
Laboratoire de Cristallographie aux Rayons X, Université de Genève, 24 quai Ernest Ansermet, CH-1211 Genève. Switzerland.
Universität Leipzig, Sektion Chemie, Wissenschaftsbereich Kristallographie, Scharnhorststr. 20, 0-7030 Leipzig. F.R.G.
University of Uppsala, Institute of Chemistry Box 531, S-751 21 Uppsala 1. Sweden.
Max-Planck-Institut für Festkörperforschung Heisenbergstraße 1, D-7000 Stuttgart 80. F.R.G.
Institut für Kristallographie und Mineralogie der Universität München, Theresienstraße 41, D-8000 München 2. F.R.G.
Institute of Crystallography, Academy of Sciences of the USSR Leninsky pr. 59, Moscow 117333. U.S.S.R.
Department of Earth Sciences, NIHON University 3-25-40 Sakurajosui, Setagayaku, Tokyo 156, Japan, Telex: Nichidai J 29496
Zeilschrift für Kristallographie
International Journal for Structural, Physical, and Chemical Aspects of Crystalline Materials
Editors-in-Chief: H. Schulz, H. G. von Schnering.
With the assistance of W. Hönle
Editorial Board: G.Bergerhoff, E.F.Bertaut, M.L.Fornasini, H.Fuess, M.Hart, P.Hartman, S.Haussühl, E.Hellner, R.J.Hill, K.Huml, Jiang Min-hua, F.Liebau, E.Parthé,
P.Paufler, S.Rundqvist, L. A. Shuvalov, Y.Takéuchi
Volume 197 1991
ρτφη R. Oldenbourg Verlag • München
Copyright It is a general condition that submitted manuscripts have not been published and will not - without written consent by the publishers - be submitted or published elsewhere. By submitting a manuscript, the authors agree that the copyright (or their article is transferred to the publisher if and when the article is accepted for publication. The copyright covers the exclusive rights to reproduce and distribute the article, including reprints, photographic reproduction, microform or any other reproductions of similar nature, and translations. Photographic reproduction, microform, or any other reproduction of text, figures, or tables from this journal is not allowed without permission obtained from the publisher.
The use of general descriptive names, trade marks etc. in this publication, even if the former are not specifically identified, is not to be taken as a sign that such names are exempt from the relevant protective laws and regulations and may accordingly be used freely by anyone. Authors are requested to present copyright releases of published materials used in their contributions.
Copyright In the USA: The appearance of the code at the top of the first page of an article in this journal indicates the copyright owner's consent that copies of the article may be made for personal or internal use, or for the personal or internal use of specific clients. This authorization to photocopy items is granted for users registered with the Copyright Clearance Center (CCC) Transactional Reporting Service, provided that the base fee of $3.00 per copy is paid directly to CCC, 27 Congress St., MA 01970. For those organizations that have been granted a photocopy license by CCC, a separate system of payment has been arranged.
The fee code for users of the Transactional Reporting Service is: 0044-2968.
This consent is given for copying beyond that permitted by Sections 107 or 108 of the U.S. Copyright Law. This consent does not extend to other kinds of copying, such as copying for general distribution, for advertising or promotional purposes, for creating new collective works or for resale.
Herstellung: Wiesbadener Graphische Betriebe GmbH, Wiesbaden
Index of Volume 197 (1991)
N u m b e r 1 / 2
W. Schwieger, K.-H. Bergk, D. Heidemann, G. Lagaly, Κ. Beneke Hochauflösende 29Si-Festkörper-NMR-Untersuchungen am syntheti-
schen Natriumsilicathydrat Makatit und dessen kristalliner Kieselsäure 1 W. A. Dolíase, C. R. Ross II
Crystal structure of orientationally disordered Na2(Ca,Sr)Si04 . . . . 13 Η. Effenberger, H. Fuess, G. Müller, T. Vogt
Crystal structure and hydrogen bonding in Li/H-exchanged petalite,
HAISÍ4O10 27 V. Busetti, G. Cevasco, G. Leandri
Crystal structure of diaryl sulphur diimides. Ortho-substituted deriva-
tives 41 A. De, G. Biswas
Crystal structure of a substituted pyranocarbazole alkaloid N-allyl
girinimbine 51 L. S. Dent G lasser, R. P. Gunawardane, R. A. Howie
The crystal structure of sodium strontium silicate, Na4SrSi309 . . . . 59 K. H. LU, C. H. Li, T. M. Chen, S. L. Wang
Synthesis and structural characterization of sodium vanadyl(IV) ortho-
phosphate NaV0P04 67
Y. Ito, S. Kuehner, S. Ghose
Phase transitions in leucite determined by high temperature, single crystal
X-ray diffraction 75 A.J.C. Wilson
Space groups rare for molecular organic structures: The arithmetic
crystal class mmmP 85 G. Paiaro, L. Pandolfo, P. Ganis, G. Valle
The crystal structure of PtíI^-írawí-Kbis-tricyclohexylphosphine)-^1^-
¿ro-formyl-íer-butylamido)(hydride)] 89 R. Basso, G. Lucchetti, A. Palenzona
Gravegliaite, MnS03 · 3 H20 , a new minerai from Val Graveglia (North-
ern Apennines, Italy) 97
IV Index of Volume 197
M. Rieder, Ζ. Weiss
Oblique-texture X-ray photographs: more information from powder
diffraction 107 U. Dettlaff- Weglikowska, E. Hey-Hawkins, D. Thiery, H. G. von Schnering
Struktur von Tetraphenylphosphonium-[(Brenzkatechin)chlorid] . . . 115 J. K. Cockcroft, A. N. Fitch
The structure of solid dichlorodifluoromethane CF2C12 by powder neu-
tron diffraction 121
Short Communication R. Dronskowski, A. Simon
Verfeinerung der Kristallstruktur von M o5G e3 131
New Crystal Structures Β. Eisenmann, A. Hofmann
Crystal structure of hexasodium di-/i-tellurido-bis(ditelluridoaluminate),
Na6Al2Te6 139
B. Eisenmann, A. Hofmann
Crystal structure of hexasodium di^-selenido-bis(diselenidoaluminate),
Na6Al2Se6 141
B. Eisenmann, A. Hofmann
Crystal structure of hexasodium di-^-thio-bis(dithiogallate) — I,
N a6G a2S6 143
B. Eisenmann, A. Hofmann
Crystal structure of hexapotassium di-/i-tellurido-bis(ditelluridogallate,
K6Ga2Te6 145
B. Eisenmann, A. Hofmann
Crystal structure of hexasodium di-/i-thio-bis(dithiogallate) — II,
N a6G a2S6 147
B. Eisenmann, A. Hofmann
Crystal structure of hexasodium di-//-selenido-bis(diselenidogallate),
Na6Ga2Se6 149
B. Eisenmann, A. Hofmann
Crystal structure of hexasodium di-^-thio-bis(dithioindate), Na6In2S6 151 B. Eisenmann, A. Hofmann
Crystal structure of hexapotassium di-^-selenido-bis(diselenidogallate),
K6Ga2Se6 153
B. Eisenmann, A. Hofmann
Crystal structure of hexapotassium di-/i-thio-bis(dithiogallate),
K6G a2S6 155
Index of Volume 197 V Β. Eisenmann, A. Hofmann
Crystal structure of hexapotassium di-/i-tellurido-bis(ditellurido-
aluminate), K6Al2Te6 157
B. Eisenmann, A. Hofmann
Crystal structure of heptasodium catenaoctaselenidotriindate(III),
Na7In3Se8 159
B. Eisenmann, A. Hofmann
Crystal structure of hexasodium di-/¿-thio-bis(dithioaluminate) — HT,
Na6Al2S6 161
Β. Eisenmann, A. Hofmann
Crystal structure of pentapotassium tetraselenidogallate(III), KsGaSe4 163 B. Eisenmann, A. Hofmann
Crystal structure of a calcium thioindate, Cao.7eIn2.84S5 165 B. Eisenmann, A. Hofmann
Crystal structure of strontium phyllotetrathiodiindate(III), SrIn2S4 . . 167 B. Eisenmann, A. Hofmann
Crystal structure of pentasodium tetrathioindate(III), Na5InS4 . . . . 169 B. Eisenmann, A. Hofmann
Crystal structure of sodium phyllo-diselenidoaluminate, NaAlSe2 . . . 171 B. Eisenmann, A. Hofmann
Crystal structure of hexapotassium di-^-selenido-bis(diselenido-
aluminate), K6Al2Se6 173
Number 3/4
Th. Kellersohn, B. Engelen, H. D. Lutz, H. Bartl, B. P. Schweiss, H. Fuess Local pseudosymmetry of the water molecule in BaBr2 · 2 H20 , a neutron
diffraction study 175 Su Genbo, Li Zhengdong
Crystal growth and physical properties of disodium subsalicylate
trihydrate 185 J. Novotny, J. Ondràcek, Β. Kratochvil, Κ. Öapek
The crystal and molecular structure of 2,3'-anhydrosucrose 189 Fan Hai-fu, H. Quan, Μ. M. Woolfson
Proteins and direct methods 197 A. Mostad, S. Natarajan
Crystal and molecular structure of the pyroelectric phase of TGS at
150 Κ 209
VI Index of Volume 197 E. Papavinasam
Crystal structure of [(bis(glycinato) copper(II)dibroniide)((bis(glycine) dilithium(I)) dihydrate)]. [((C2H4N02)2CuBr2) • ( ( C2H5N 02)2L i2) • 2
H20 ] 217
L. Born, G. Heywang
The crystal structure of the radical anion salt of naphthalene tetra- carboxylic dianhydride (NTDA) and 5,6-dihydro-4a,6a-phenanthrolene
dionium (DPD) : a new charge transfer complex 223 L. Eriksson, P. Wang, P.-E. Werner
Crystal structure and phase transition of a-Cu2HgI4 235
New Crystal Structures
M. Jansen, G. Q. Wu, K. Königstein
Crystal structure of caesium ytterbium diphosphate, C s Y b P207 . . . 245 B. Liier, M. Jansen
Crystal structure refinement of tetraphosphorous nonaoxide, P409 . . 247 M. Somer, K. Peters, H. G. von Schnering
Crystal structure of caesium potassium rubidium 2,5-diantimonido-tristi-
ba-2,5-digalla-cyclopentanate, C s3.31K2.2 5R b144G a2S b5 249 B. Eisenmann, J. Jäger
Crystal structure of caesium catena-tritelluroaluminate, CsAlTe3 . . . 251 B. Eisenmann, J. Jäger
Crystal structure of hexacaesium di^-telluro-bis(ditelluroaluminate),
Cs6Al2Te6 253
B. Eisenmann, R. Zagler
Crystal structure of tripotassium tritelluroantimonate(III), K3SbTe3 . 255 B. Eisenmann, R. Zagler
Crystal structure of tripotassium tritellurobismutate(III), K3BiTe3 . . 257 B. Eisenmann, J. Jäger, R. Zagler
Crystal structure of tetrapotassium tetratellurodiarsenate(II), K4As2Te4 259 B. Eisenmann, R. Zagler
Crystal structure of tetrasodium tetratellurodiarsenate(II), Na4As2Te4 261 B. Eisenmann, J. Klein, M. Somer
Crystal structure of pentarubidium triphosphidogermanate, R b5G e P3 263 B. Eisenmann, J. Klein, M. Somer
Crystal structure of decasodium di-/i-arsenido-bis(diarsenidogermanate),
NaioGe2As6 265
B. Eisenmann, J. Klein, M. Somer
Crystal structure of decasodium di-^-arsenido-bis(diarsenidosilicate),
NaioSi2As6 267
Index of Volume 197 ν π Β. Eisenmann, J. Klein, M. Somer
Crystal structure of decasodium di-/i-phosphido-bis(diphosphido-
stannate(IV)), Na10Sn2P6 269
B. Eisenmann, J. Klein, M. Somer
Crystal structure of tetrapotassium diarsenidocadmate, K4CdAs2 . . . 271 B. Eisenmann, J. Klein, M. Somer
Crystal structure of pentarubidium triarsenidosilicate, Rb5SiAs3 . . . 273 B. Eisenmann, J. Klein, M. Somer
Crystal structure of trisodium dipotassium triphosphidosilicate,
Na3K2SiP3 275
B. Eisenmann, J. Klein, M. Somer
Crystal structure of dipotassium catena-phosphidoaurate(I), K2AuP . 277 B. Eisenmann, J. Klein
Crystal structure of tetrasodium monopotassium triarsenidogermanate,
Na4KGeAs3 279
G. Cordier, H. Ochmann
Crystal structure of disodium phyllo-triantimonidodiindate, Na2In2Sb3 281 G. Cordier, H. Ochmann
Crystal structure of disodium phyllo-triarsenidodialuminate,
Na2Al2As3 283
G. Cordier, H. Ochmann
Crystal structure of disodium phyllo-triarsenidodigallate, Na2Ga2As3 285 G. Cordier, H. Ochmann
Crystal structure of dipotassium phyllo-triarsenidodigallate, K2Ga2As3 287 G. Cordier, H. Ochmann
Crystal structure of dipotassium phyllo-triantimonidodigallate,
K2Ga2Sb3 289
G. Cordier, H. Ochmann
Crystal structure of dipotassium phyllo-triantimonidodiindate,
K2In2Sb3 291
G. Cordier, H. Ochmann
Crystal structure of dipotassium phyllo-triarsenidodiindate, K2In2As3 293 G. Cordier, H. Ochmann
Crystal structure of tripotassium phyllo-triarsenidoindate, K3In2As3 295 G. Cordier, H. Ochmann
Crystal structure of potassium phyllo-diantimonidogallate, KGaSb2 . 297 M. Nieger, O. Altmeyer, E. Niecke
Crystal structure of l,2-bis-[tris-(trimethylsilyl)hydrazino]-l,2-dibromo-
diphosphane, (((CH3)3Si)3N2(Br)P)2 299
V i l i Index of Volume 19
F. Knoch
Crystal structure of 8-(2-hydroxyethoxy)quinoline dihydrate,
C9H6N0CH2CH20H(H20)2 301
W. P. Bosman, P. T. Beurskens, G. Admiraal, R. P. Sijbesma, R. J. M.
Nolte
Crystal structure of l,6:3,4-bis(4,5-benzo-3,6-dimethoxy-l,2-xylylene) tetrahydro - 3a,6a - diphenylimidazo [4,5-d] imidazole - 2,5(1H,3H) - dione,
C44H38N406 3Œ
K. Peters, E.-M. Peters, H. G. vort Schnering, W. Adam, S. E. Bottle Crystal structure of 2,4,4-triphenyl-2-aza-3-oxabicyclo[3.3.0]oct-5-ene,
C2 4H2 1NO 30)
G. Cordier, C. Röhr
Crystal structure of strontium gold gallium (1/2/2), SrAu2Ga2 . . . . 312 G. Cordier, C. Röhr
Crystal structure of calcium copper indium (1/4/1), CaCu4In 314 Book Reviews
W. Borchardt-Ott
Kristallographie. 3. Auflage. (Rez. : J. Böhm) 317 R. Vanselow, R. Howe (Eds.)
Chemistry and Physics of Solid Surfaces VIII. (Rez. : A. Meisel) . . . 31«
R. Iffländer
Festkörperlaser zur Materialbearbeitung. (Rez.: W. Pompe) 318 A. R. Holzel
Systematics of Minerals. (Rez.: H.-J. Hobler) 319 H. Zabel, S. A. Solin (Eds.)
Graphite Intercalation Compounds I. (Rez. : H. Eschrig) 320 A. Lösche
Molekulare Ordnung und Orientierung, insbesondere bei Flüssigkristal-
len. (Rez.: D. Demus) 321 C. B. Harris, E. P. Ippen, G. Α. M our ou, Α. Η. Zewail (Eds.)
Ultrafast Phenomena VII. (Rez.: Κ. Kreher) 322
Author Index of Volume 197 323 Subject Index of Volume 197 325 Formulae Index of Volume 197 325
Zeitschrift für Kristallographie 197, 1 - 1 2 (1991)
© by R. Oldenbourg Verlag, München 1991 - 0044-2968/91 $ 3.00 + 0.00
Hochauflösende
2 9
Si-Festkörper-NMR-Untersuchungen
am synthetischen Natriumsilicathydrat Makatit und dessen kristalliner Kieselsäure
Wilhelm Schwieger und Karl-Heinz Bergk*
Sektion Chemie der Martin-Luther-Universität Halle-Wittenberg, Schloßberg 2, 0-4020 Halle/Saale
Detlef Heidemann
Zentralinstitut für Anorganische Chemie der Akademie der Wissenschaften, Rudower Chaussee 5, 0-1199 Berlin-Adlershof
Gerhard Lagaly und Klaus Beneke
Institut für Anorganische Chemie der Christian-Albrechts-Universität Kiel, Olshausenstraße 40/60, W-2300 Kiel
Eingegangen am 23. Oktober 1989; angenommen am 7. Januar 1991
Sodium silicate hydrates / Crystalline silicic acids / 2 9 Si-MA S-NMR-spectra
Abstract." Solid-state high-resolution 29Si-NMR experiments were used to study the structure of the silicate layers in makatite and its crystalline silicic acid obtained by a cation exchange reaction from makatite. For each of the studied samples two well-separated NMR signals have been recorded which could be assigned to Q3 — [—OSi(OSis)3] — units of the silicate layers of the corresponding compounds and to Q4 — [Si(OSi Ξ )4] — units of quartz impurities. Compared with makatite in the silicic acid a high-field shifted g3-signal and a splitting of this signal into two component lines have been detected. These observations were attributed to structural changes which appeared as a result of the cation exchange reaction.
* Sonderdruckanfordenmgen an Prof. Dr. K.-H. Bergk.
2 W. Schwieger et al.
Einleitung
1970 wurde von Sheppard, Gude, Hay (1970) das Natriumsilicatmineral Makatit beschrieben, das von Hay (1968) in den Sedimenten des Sees Magadi (Kenia) gefunden wurde.
Makatit ist der Gruppe der Natriumsilicathydrate mit Schichtstruktur zuzuordnen. Zu diesen zählen auch Ilerit1, Magadiit und Kenyait. Der Makatit stellt, betrachtet man den Kondensationsgrad ausgedrückt als Si02/Na20-Molverhältnis, eines der Anfangsglieder dieser Gruppe dar (Schwieger, Heyer, Wolf und Bergk, 1987; Bergk, Schwieger und Porsch, 1987).
Erste Synthesen wurden von Lagaly (1979) und Beneke und Lagaly (1984) beschrieben. Eine umfassende Charakterisierung der Reaktionsfä- higkeit des Makatits liegt bisher nicht vor. Annehed, Fälth und Lincoln (1982) gelang es, Makatitkristalle hydrothermal zu synthetisieren, die eine Röntgeneinkristallstrukturanalyse erlaubten. Nach diesen Untersuchungen besteht Makatit aus Silicateinfachschichten mit einer Identitätsperiode von 4 Si04-Tetraedern — also einer 4er Einfachschicht. Der resultierende Anio- nenkomplex läßt sich formal als (Si4O10)4~ formulieren. Aus diesem Auf- bau ergibt sich eine Anordnung der Si04-Tetraeder, die, folgt man der Q"- Klassifikation [Lippmaa et al. (1980)], nur ß3-Gruppen enthält. NMR- Untersuchungen an Natriumsilicathydraten bzw. deren Kieselsäuren wur- den bisher von vier Arbeitsgruppen vorgestellt.
Wittich, Voigtländer und Lagaly (1975) wiesen durch Messung der transversalen Relaxationszeit in unterschiedlichen Proben des syntheti- schen H-Magadiits sowie in einem synthetischen Magadiit Protonen von SiOH-Gruppen und H20-Molekülen nach und diskutierten das aus den Relaxationszeiten bestimmte Verhältnis der unterschiedlich gebundenen Protonen.
Rojo, Ruiz-Hitzky, Sanz und Seratosa (1983) charakterisierten mittels Protonenresonanzuntersuchungen an H-Magadiiten unterschiedlicher Her- kunft die Art der SiOH-Gruppen und leiteten aus 29Si-MAS-NMR-Unter- suchungen (Rojo, Sanz, Ruiz-Hitzky und Seratosa 1986) das Modell einer Zweifachschicht zur Beschreibung der Silicatschichten im H-Magadiit ab.
Ausgehend von 29Si-NMR-Messungen stellten Pinnavaia, Johnson und Lipsicas (1986) ein ähnliches Modell mit Tetraederdoppelschichten für den Magadiit und seine Kieselsäure auf. Schwieger et al. (1985) und Heidemann, Schwieger und Bergk (1987) führten systematische Untersuchungen an den synthetischen Natriumsilicathydraten Ilerit, Magadiit und Kenyait und deren Kieselsäuren zur Strukturaufklärung durch. Insbesondere durch 29Si- NMR-Messungen konnten Aussagen über systematische Veränderungen in
1 Die Bezeichnung „Ilerit" wurde von uns analog der Bezeichnung in einer früheren Arbeit (Wolf et al. 1979) für ein Silicat nach Her (1964) verwendet. Wir sind uns bewußt, daß sie streng genommen nicht gültig ist und auf ein Mineral hindeuten würde, was nicht zutrifft.
29Si-MAS-NMR-Untersuchungen am Makatit und dessen Kieselsäure 3 der Silicatschicht in Abhängigkeit von der Art des Schichtsilicates und seines Si02/Na20-Molverhältnisses getroffen und ein Strukturmodell abge- leitet werden, das die Silicatschichten im Ilerit und seiner Η-Form als Zweifachschichten, die im Magadiit und seiner Η-Form als Dreifachschich- ten und die im Kenyait und H-Kenyait als Fünffachschichten beschreibt.
In der vorliegenden Arbeit wird über Untersuchungen zur Strukturcha- rakterisierung eines synthetischen Makatits und dessen kristalliner Kiesel- säure berichtet, um eine Einordnung entsprechend ihrer Si02/Na20-Mol- verhältnisse von 4 in die Substanzreihe Ilerit (Si02/Na20 = 8), Magadiit (Si02/Na20 = 14) und Kenyait (Si02/Na20 = 20) und deren Kieselsäuren vornehmen zu können.
Experimentelles
SyntheseZur Herstellung des Makatits wurde festes Si02 (Kieselsäure, gefallt, Merck) in wäßriger Natronlauge dispergiert und bei 100°C hydrothermal umgesetzt. Das molare Ansatzverhältnis betrug 3,6 Si02/2,0 NaOH/
13,3 H20 . Das Kristallisat besaß nach dem Auswaschen eine Zusammenset- zung von 1,08 N a20 · 4 Si02 · 5,17 H20, die dem Literaturwert von Na2Si409 · 5 H20 nahekommt. Die Herstellung des H-Makatits — der resultierenden kristallinen Kieselsäure — erfolgte durch Elution der Na- triumionen mit 1 η HCl-Lösung bei Raumtemperatur und einem fest/flüs- sig-Verhältnis von 1/100.
Charakterisierungsmethoden
Die Röntgendiffraktogramme wurden mit einem Vertikal-Zählrohr-Gonio- meter (Cu—Ä^-Strahlung, Ni-Filter, 36 kV, 24 mA) aufgenommen. Als in- nerer Standard diente Silicium mit dem Reflex bei 0,3137 nm. Die elektro- nenmikroskopischen Untersuchungen erfolgten an einem Elektronen- mikroskop JEM 100 S der Firma JOEL in Rastertechnik. Die Proben wurden kurzzeitig in wäßriger Lösung mittels Ultraschall suspendiert und nach dem Auftragen und Trocknen mit Kupfer und Kohlenstoff im Hochvakuum bedampft.
Die NMR-Untersuchungen wurden bei 12,03 MHz (Eisenmagnet, 1,4 T) am Vielimpuls-Festkörper-NMR-Spektrometer FKS 176/178 durch- geführt. Als Nachweistechniken wurden die konventionelle FT-Technik bei gleichzeitiger Protonenentkopplung und das Kreuzpolarisationsverfahren (CP), beides in Kombination mit schneller Rotation der Probe um den magischen Winkel (MAS), eingesetzt. Die CP-Technik erfaßt alle Si-Tetra- eder, die eine Kopplung zu Protonen besitzen. Es wurde festes [(CH3)3Si]8Si802o (M8Q8) als sekundärer Standard verwendet, dessen M- Signal auf 11,5 ppm gesetzt wurde. Alle Werte der chemischen 29Si-Ver-
4 W. Schwieger et al.
ID Ν S ce H 3 c/3 a <
fS
o* <N
o"
« t : . 0) IE =5
r- -o ON 00
ι no
o o "
r^ >o m m m cn oo o" o" o O
ON t- On <r\ Ci O
w, NO ON «O m Ν 0C
O NO
ΓΟ CN| <N <N fN (N tN O* o" o" o ' O o o* o"
i r j m o m *o Tt ON ιλ m ~ ο Ό (Ν O OO
« (Ν
Λ ai ' Os 00 Tt Tt^ τ* co rn ro en ro O O O O O O O O Ö" o o " o
OO NO OO » n C— O V ï V I (N
<N On OO r - CO NTi on V Ì NO On OO t-NO NO *r> tJ-rr
O l <N fN <N <N <N (N tN <N
O o O o" O* O o o " o o"
r i o\ o ^ ^ σ\
ι^ (Ν o rs
w U·
~ s « s· fi·
ce jjj S on
ε
•y
no o r-
^ Ν σ\ OO Ν « ο τ»· o t- v> γν|
ON oo »Λ Tj· Tt TÍ·
o " o " o o o" o ' rncncoeoenf^fnrncN<N<N (N c^
Ο Ο* Ο o* o" o" o ' o" o ' o " o" o o"
O m « n ON
•Π <N CN|
o o ' Τ NO
00 V I
m m
<N <N
o o •t
r - o m τ - Ν m - O O N I N O Ο Ν M " O m
VN-^-fNOOVl^CINOVÌOCNl-^- <vn Tj- o m
(S Tf (S T-
nO NO
29Si-MAS-NMR-Untersuchungen am Makatit und dessen Kieselsäure 5
<N r—
oo m
<N <S
<N rs θ" O
00 O (Ν OO Os
<N -η
rtOOVOOíS^fnmxt^
N P K S t h w O O V O O O O O O
^ N. "*. "1 *". * ΐ o o o o o o o o o o
fO Vi Ό Γ+ »Λ --»-i Ο 3 ν 0 0 \ · ψ Ν Ι »
^ O O V C O U Q O h Ή Ή T. 1 1 " i *"ί o o" o o o o o
so so (N O
oo o 2 S OS <N o o"
Γ<1 Vi
0\ Tf 00 00
so so O «o O m Tf O so t so 00
6 W. Schwieger et al.
Schiebung sind auf Tetramethylsilan (TMS) bezogen und in der ¿-Skala angegeben. Die Reproduzierbarkeit der gemessenen isotropen Verschie- bungswerte war 1 ppm.
Ergebnisse und Diskussion
Tabelle 1 enthält die beobachteten Röntgenreflexe der Makatitproben. Die Werte sind den Angaben von Sheppard et al. (1970) für den natürlichen Makatit gegenübergestellt. Die im Rahmen der Fehlergenauigkeit liegende Übereinstimmung der Linienlagen weist unser Produkt als Makatit aus.
Dies wird auch durch die Intensitätsabstufung der Reflexe unterstützt. Eine Optimierung der Gitterkonstanten auf Basis der Indizierung von Sheppard wurde von uns bisher nicht vorgenommen. Für den H-Makatit sind Ver- gleichsdaten aus der Literatur nicht bekannt.
Der Kationenaustauschprozeß ist reversibel und führt wieder zum Aus- gangsmakatit mit identischen Linienlagen. Die Reflexe bei 0,426; 0,3343 und 0,246 nm, die sowohl im Spektrum des Makatit als auch des H-Makatit beobachtet werden, sind Quarz zuzuordnen und zeigen Quarzanteile in den Proben an (vergi. Tabelle 1). Dieser Befund wird durch die 29Si-MAS- NMR-Untersuchungen bestätigt. Die Quarzanteile sind auf den REM- Aufnahmen, Abbildung 1, nicht erkennbar. Die Aufnahmen weisen den Makatit in der Syntheseform als Kristallite mit faseriger Morphologie aus.
Dies steht damit im Kontrast zu dem vorwiegend blättrigen schichtartigen Erscheinungsbild der anderen Natriumsilicathydrate, wie Ilerit, Magadiit und Kenyait.
Die Ergebnisse der 29Si-MAS-NMR-Untersuchungen von Makatit und H-Makatit sind in Abbildung 2 dargestellt. Es werden für beide Verbindun- gen das ^-entkoppelte FT-MAS-Spektrum (oben) und das entsprechende CP-MAS-Spektrum (unten) gezeigt. Im FT-Spektrum werden zwei Signale beobachtet. Das Signal bei —93,6 ppm für den Makatit und —102,7 ppm für H-Makatit ist in Übereinstimmung mit Literaturdaten 03-Baugruppen, das Signal bei —107,5 ppm Ö4-Baugruppen zuzuordnen (Lippmaa et al., 1980; Mägi et al., 1984; Smith und Blackwell, 1983). Das Auftreten eines ö"-Signals in der Makatitprobe ist überraschend.
. Entsprechend den Strukturdaten Anneheds et al. (1982) sollten nur Q3- Baugruppen zu beobachten sein. Da im CP-MAS-Spektrum sowohl für Makatit als auch für H-Makatit kein ß4-Signal erscheint, läßt dies den Schluß zu, daß die Q4-Signale der FT-Spektren anderen kristallinen Phasen zuzuordnen sind. Diese Aussage wird auch durch die unveränderte Lage des <24-Signals vor und nach dem Ionenaustausch unterstützt, da frühere Untersuchungen an anderen Natriumsilicathydraten auch eine Beeinflus- sung des ß4-Signals nachwiesen. Der Wert von —107,5 ppm ist identisch mit dem Signal, das für α-Quarz angegeben wird (Lippmaa et al. (1980);
29Si-MAS-NMR-Untersuchungen am Makatit und dessen Kieselsäure 7
Abb. 1. Rasterelektronenmikroskopische Aufnahmen des synthetischen Makatits.
Smith et al. (1983)). Es muß deshalb eine Verunreinigung der Probe durch Quarz angenommen werden. Diese Aussage ist in Übereinstimmung mit den Ergebnissen der röntgenografischen Charakterisierung der untersuch- ten Proben.
Nicht unterschieden werden kann, ob diese Verunreinigung synthesebe- dingt auf Grund von Rekristallisationserscheinungen oder durch eine Alte- rungsreaktion bei der Lagerung des Produktes entstanden ist. Dennoch deutet das Auftreten des Quarzes an, daß auch der Makatit — ebenso wie alle anderen Natriumsilicathydrate — eine metastabile Phase im Quarzbil- dungsprozeß darstellt.
Die Strukturierung des Ö3-Signals im FT-Spektrum sowohl des Maka- tits als auch seiner Kieselsäure weist darauf hin, daß die Silicatschichten in beiden Verbindungen aus kristallograflsch nicht identischen Si04-Tetra-
8 W. Schwieger et al.
MRKHTIT H-MRKflTIT
-93. E -I07.S
-102.7 FT-5PEKTREN
-ID2.7
IDO.2I
CP-5PEKTREN
W v v ^
-90 -120 -90 -120 <f(ppm) Abb. 2.12 M H z 2 9Si-MAS-NMR-Spektren des synthetischen Makatits und seiner kristal--Sä-
linen Kieselsäure.
edera aufgebaut sind. Für den Makatit steht dies in Übereinstimmung mit den von Annehed et al. (1982) ermittelten Strukturdaten. Eine Aufspaltung des Ö3-Signals ist durch Messung bei erhöhten Feldstärken zu erwarten.
Eine Korrelation zwischen den NMR-Ergebnissen und den Strukturdaten ist dann geplant.
Die Spektren des H-Makatits weisen im Vergleich zum Ausgangssilicat bezüglich ihrer <23-Signale eine Hochfeldverschiebung auf, wie sie auch für andere kristalline Kieselsäuren gegenüber ihren Ausgangsverbindungen beobachtet wird (Heidemann et al. (1985), (1987)).
In Tabelle 2 ist dies vergleichend dargestellt. Auffallend ist, daß der Betrag der Verschiebungsdifferenz der Ö3-Signale von Ausgangssilicat und entsprechender Kieselsäure stark differiert und offensichtlich von der Struktur der Silicatschicht abhängt.
Mit dem Übergang von Silicateinfach- zu Silicatmehrfachschichten ver- ringert sich der Betrag der Hochfeldverschiebung systematisch.
2 9S i - M A S - N M R - U n t e r s u c h u n g e n a m M a k a t i t u n d d e s s e n K i e s e l s ä u r e 9
<
3T J G 3
οο ^ NO <N J> — ON ^ , OO — « ' i On . C w w N gO o w — r¡ „ ο υ
" 3 ~ 2 ' S Î 3 S í S - S 5 c
OH h j C <
ω
"S o
Ν
2 ? *** οο . On S
NO 00 on
f- b»
u »
to 00 J= .H O > « >
« s
O « 7 3 2 S -
" M U
" St fi
<2 JG rt
* η μ g ( Λ β Û
w  w (t J 3 " J= — . B *>
l e - g 2 g βί « j ^ ^
• C U . f i >
u . υ , 8 . 2 . a î s ? e
°e6
Ν
w oo ON 7 3 ·» w
£ w ι • . a u - 6 «
t a l «
• S - S - S - s
¡ I I I . S c o S β fc
>
Ν Ό c ι
SA
s ¡ I
<N
o I
• c
α S "
en Ü O ) υ
X ) ω 3 I S IH
£ I c/3
c o «
« s
ed eo M 3
< I ζ
f i OV o
• a :cS Λ ! Id ¡a
o •-ι
O O
7
»r> y—s Ό
0 0 0 0 0 0 ΟΟ
ON ON On On 1
— ' ' — ' *—'
" 3 Td 7 3 " 3
«-» •4-t
υ υ υ
Η tH cd ι*
ν υ υ
00 οο " 3 οο
υ ' S « > υ
' ! :·5 ξ • ο cd e ' S * Λ ο cd J 3 e o υ cd c οο cd e o
"Π co cd (Λ οο cd ε S 1 / 3
S ι ι s
c υ
•fi E υ
"O ν
>ο ο
Ι - , (Ν CS ο - 8 S
00 ο
7
00, οο*
ο\
I
υ Ο.
Ο
£
NO ο ο on
:cd 3 «9
« £ (Ο Η . 2 M
Ζ
— «β 7 > | ο - 8
• J u
" f t a s κ
Μ cd cd
Κ
S οο l e .
"C 12 JO o
•τ* Κ o
— s 0 0 c ρ : s g o\
· " T3 Ë «
w cd Ö '
— S P O — cd « ·δ cd
« »
ao ocd Κ
- o
w « ΟΟ H3 ' f ~
S , « — SP c —·
• S - S 2
~ 0L| «
C p S 00
« s »
• R i » o 53 « Κ S
10 W. Schwieger et al.
Nach Heidemann et al. (1987) leitet sich diese Hochfeldverschiebung des ß3-Signals für die Kieselsäuren direkt aus der Veränderung charakteri- stischer Strukturparameter der Si04-Tetraeder — des mittleren Si —O- Abstandes d (Si — O ) und des über die drei Si — O — Si-Bindungen gemittelten Bindungswinkels SiOSi — beim Übergang vom Natriumsili- cat zur entsprechenden Kieselsäure ab. Diese Parameter sind sowohl für das Natriumphyllosilicat a-Na2Si205:[á(Si —O)] = 0,1617 nm und
-fc SiOSi = 146° (Pant et al., 1968) als auch für die entsprechende Kiesel- säure [Daten der von Liebau (1964) als „idealisierte Struktur" bezeichneten Strukturvariante: d ( S i - O ) = 0,161 nm und * SiOSi = 155°] bekannt. Es ist ersichtlich, daß als entscheidende Veränderung der deutlich vergrößerte mittlere Bindungswinkel der ß3-Baugruppen anzusehen ist. Folgt man den Überlegungen Heidemanns et al. (1987), so wird die Möglichkeit der Winkelvergrößerung beim Übergang von Silicateinfachschichten zu Silicat- mehrfachschichten eingeschränkt, d. h. deren Beweglichkeit behindert. Dies kommt in der Verringerung der Verschiebungsdifferenzen der Ö3-Signale zum Ausdruck und wird verständlich, wenn man die hohen (^-Gruppen- Anteile in den Verbindungen mit Silicatmehrfachschichten berücksichtigt (Schwieger et al. (1985)).
Die Werte für Makatit und H-Makatit ordnen sich demzufolge gut in diese Abstufung ein.
Beim Übergang vom Makatit zum H-Makatit ändert sich jedoch der isotrope Wert der chemischen 29Si-Verschiebung um 9,1 ppm gegenüber 7,2 ppm bei der Umwandlung von a-Na2Si205 in H2Si205-l (s. Tabelle 1).
Die größere Hochfeldverschiebung weist auf eine höhere Flexibilität der Silicatschichten des Makatits beim bzw. nach dem Protonenaustauschvor- gang hin. Dies kann möglicherweise mit den unterschiedlichen Identitäts- perioden der Si04-Tetraeder der Silicatschichten in Makatit (4er-Einfach- kette in der Schicht) und in a-Na2Si205 (2er-Einfachkette in der Schicht) erklärbar sein.
Neben der Hochfeldverschiebung des (?3-Signals für den H-Makatit wird außerdem eine Aufspaltung dieses Signals gegenüber dem entsprechen- den Signal im Makatit beobachtet. Während im FT-Spektrum nur eine Schulter bei —100 ppm daraufhindeutet, wird im CP-Spektrum eine deutli- che Aufspaltung in zwei Einzelsignale bei —100,2 und —102,7 ppm nachge- wiesen. Dieser Befund weist zweifelsfrei auf die Existenz von mindestens zwei strukturell deutlich verschiedenen Si04-Tetraedern in der Silicat- schicht des H-Makatits hin.
Aus Tabelle 2 ist ersichtlich, daß Aufspaltungen der ß3-Signale für die Natriumsilicathydrate und deren Kieselsäuren bisher nicht beobachtet wurden.
Die hier vorgestellten und diskutierten Ergebnisse der 29Si-M A S - N M R - Untersuchungen erlauben somit den Schluß, daß mit der Überführung des Makatits in seine Kieselsäure strukturelle Veränderungen der Si04-
"Si-MAS-NMR-Untersuchungen am Makatit und dessen Kieselsäure 11
Tetraeder in der Silicatschicht einhergehen. Wie bereits für andere Natrium- silicathydrate diskutiert (Heidemann et al. (1987)), deutet sich auch für die Si04-Tetraeder der Makatitschicht im Ergebnis des Kationenaustausches eine Vergrößerung des mittleren Si—O —Si-Bindungswinkels an.
Die Schulter des ß4-Signals bei — 114ppm im FT-Spektrum des H- Makatits deutet, wie auch für andere Natriumsilicathydrate beobachtet (Heidemann et al. (1987)), auf eine Erhöhung des ß4-Gruppen- Anteils nach dem Protonenaustausch auch für Makatit und dessen Protonenform hin.
Eine Zuordnung zum Quarz ist auszuschließen, da, wie oben beschrieben, Quarz eine Linie bei —107,5 ppm aufweist. Das Signal bei — 114 ppm läßt eine Kondensation zwischen ß3-Gruppen benachbarter Makatitschichten vermuten. Der hohe Wert der Verschiebung deutet auf stark verzerrte Bindungen hin.
Für die Anfertigung der REM-Aufnahmen gilt unser Dank Herrn R. Schneider vom Zentralen Forschungsinstitut für Elektronenmikroskopie der Akademie der Wissenschaf- ten, Halle.
Literatur
Annehed, H., Fälth, L., Lincoln, F. J.: Crystalstructure of synthetic makatite Na2Si408(0H)2 · 4 H20 . Z. KristaUogr. 159 (1982) 203 - 210.
Beneke, K., Lagaly, G.: Kristalline Kieselsäuren. GIT Fachz. Lab. 28 (1984) 512 - 527.
Bergk, K.-H., Schwieger, W., Porsch, M. : Aluminiumfreie Schichtsilicathydrate. Synthese und Eigenschafts-Anwendungs-Beziehungen Teil II: Die Struktur der MetaUsilicat- hydrate M —SH und ihre Eigenschafts-Anwendungs-Beziehungen. Chem. Tech. 39 (1987)508-514.
Brandt, Α., Schwieger, W., Bergk, K.-H.: A new model structure of sheet sodium (Na) silicate hydrates (Na—SH) — theoretical view based on known X-ray and NMR- measurements. Rev. Chim. Miner. 24 (1987) 5 6 4 - 571.
Hay, L. R. : Chert and its sodium-silicate precursors in sodium-carbonate lakes of East Afrika. Contrib. Mineral. Petrol. 17 (1968) 255 - 274.
Heidemann, D., Grimmer, A.-R., Hübert, C., Starke, P., Mägi, M. : Hochauflösende 29Si- Festkörper-NMR-Untersuchungen an polykristallinen Phyllokieselsäuren (H2S12O3) bei tiefem und hohem Magnetfeld. Z. Anorg. Allg. Chem. 528 (1985) 2 2 - 36.
Heidemann, D., Schwieger, W., Bergk, K.-H.: Hochauflösende "Si-Festkörper-NMR- Untersuchungen an synthetischen siliciumreichen Kieselsäurehydraten. Z. Anorg.
Allg. Chem. 555 (1987) 129-142.
Her, R. K.: Ion exchange properties of a crystalline hydrated silica. J. Colloid Sci. 19 (1964)648 - 657.
Lagaly, G.: Crystalline silicic acids and their interface reactions. Adv. Colloid Interface Sci. 11 (1979) 105-148.
Liebau, F.: Über die Kristallstrukturen zweier Phyllokieselsäuren H2SÌ2O5. Z. Kristal- logr. 120 (1964) 427 -449.
Lippmaa, E., Mägi, M., Samoson, Α., Engelhardt, G., Grimmer, A.-R.: Structural studies of silicates by solid-state high-resolution 29Si NMR. J. Am. Chem. Soc. 102 (1980) 4889-4893.
Mägi, M., Lippmaa, E., Samoson, Α., Engelhardt, G., Grimmer, A.-R. : Solid-state high- resolution silicon-29 chemical shifts in silicates. J. Phys. Chem. 88 (1984) 1518-1522.
12 W. Schwieger et al.
Pant, Α. Κ., Cniickshank, D. W. J. : The crystal structure of α N a2S i205. Acta Crystallogr.
Β24 (1968) 1 3 - 1 9 .
Pinnavaia, T. J., Johnson, I. D., Lipsicas, M.: A 29Si M A S - N M R study of tetrahedral site distributions in the layered silicic acid H+-magadiite ( H2S i1 4029 · η H20 ) and in N a+- m a g a d i i t e ( N a2S i1 402 9 · η H20 ) . J. Solid State Chem. 63 (1986) 1 1 8 - 1 2 1 . Rojo, J. M., Ruiz-Hitzky, E., Sanz, J., Seratosa, J. M.: Characterization of surface Si —
O H groups in layer silicic acids by IR and N M R spectroscopies. Rev. Chim. Miner.
20(1983) 8 0 7 - 8 1 6 .
Rojo, J.M., Sanz, J., Ruiz-Hitzky, E., Seratosa, J. M. : 2 9Si M A S - N M R spectra oflamellar silicic acid H-magadiite and its trimethylsilyl derivative. Z. Anorg. Allg. Chem. 540/
541 (1986) 227.
Schwieger, W., Heidemann, D., Bergk, K.-H.: High-resolution solid-state silicon-29 nuclear magnetic resonance spectroscopic studies of synthetic sodium silicate hy- drates. Rev. Chim. Miner. 22 (1985) 6 3 9 - 6 5 0 .
Schwieger, W., Heyer, W., Wolf, F., Bergk, K.-H.: Zur Synthese von kristallinen Metall- silicathydraten mit Schichtstruktur. Z. Anorg. Allg. Chem. 548 (1987) 204 - 216.
Sheppard, A. R., Gude, A. J., Hay, L. R.: Makatite — a new hydrous sodium silicate mineral from Lake Magadi, Kenya. Am. Mineral. 55 (1970) 358 — 366.
Smith, J. V., Blackwell, C. S. : Nuclear magnetic resonance of silicate polymorphs. Nature (London) 303 (1983) 2 2 3 - 2 2 5 .
Wittich, E. K.-H., Voigtländer, J., Lagaly, G. : Kernmagnetische Relaxationszeit-Messun- gen an OH-Protonen einer kristallinen Kieselsäure. Ζ. Naturforsch. 30a (1975) 1 3 3 0 - 1 3 3 1 .
Wolf, F., Schwieger, W.: Zum Ionenaustausch einwertiger Kationen an synthetischen Natriumpolysilicaten mit Schichtstruktur. Z. Anorg. Allg. Chem. 457 (1979) 2 2 4 - 228.
Zeitschrift für Kristallographie 197, 1 3 - 2 6 (1991)
© by R. Oldenbourg Verlag, München 1991 - 0044-2968/91 $ 3.00 + 0.00
Crystal structure
of orientationally disordered Na
2(Ca,Sr)Si0
4 W. A. Dolíase1·2 and C. R. Ross II21 Department of Earth and Space Sciences, University of California, Los Angeles, California, USA
2 Bayerisches Geoinstitut, Universität Bayreuth, W-8580 Bayreuth, Federal Republic of Germany
Received: December 6,1989; revised August 30,1990
Orientational disorder / X-ray powder diffraction / Cubic harmonics / Sodium calcium silicate / Sodium strontium silicate
Abstract. Na2CaSi04, Na2SrSi04, and Na2(Cao.5Sro.5)Si04, are shown by X-ray powder diffraction to have the cubic, Fm3m crystal structure of orientationally disordered Na3P04. The distribution of Si04 orientations are well described by cubic harmonics and show a high probability of Si—O bonds pointing within about 20° of <100> and low probabilities in other directions. The disordered Si04 groups form a cubic close packed array with cations in both "tetrahedral" and "octahedral" interstices. There is apparent preference of the divalent Ca and Sr cations for the tetrahedral sites over the octahedral sites. About 92% of the cations in the ideal formulae are accounted for on these two sites. Electron microprobe analyses suggest that at least part of any remaining deficit is attributable to loss of Na atoms during synthesis but additional, weakly occupied sites may also be present in the structure.
Introduction
Orientationally disordered crystals (ODC's) are compounds in which the orientation of some atomic group is not periodically repeated. The subject has been reviewed by Parsonage, Stavely (1978). The variety of orientations adopted by the group results in a variety of local bonding configurations
Correspondence: W. A. Dolíase, Department of Earth and Space Sciences, UCLA, Los Angeles, CA 90024, USA.
14 W. A. Dolíase and C. R. Ross II
between the outermost atoms of the group and its surrounding shell of atoms. Formation of O D C ' s is favored by these bonds being weak — conditions met in particular when both the atomic group and its coordinating atoms are low-valent and have high coordination numbers.
The disordered groups themselves tend to be tightly bonded, rigid and roughly globular in shape ; common examples are C N " , N H 4 , C O | ~, SO4 "
and PO4 ~. The surrounding atoms are typically large alkali, alkaline-earth or halogen elements.
O D C structures are transitional between the normal, ordered crystalline state and the orientationally and translationally disordered liquid or glassy state. Consequently, their physical properties tend to be intermediate be- tween these end member states of matter, commonly showing anomalous thermal, transport, solid-solubility and mechanical properties. The several discrete orientations present in O D C ' s are separated by an energy barrier which is occasionally surmounted resulting in a temperature-dependent reorientation rate (Press, 1981). With decreased temperature (or in some cases increased pressure) O D C ' s usually transform to orientationally ordered structures, with the phase change not uncommonly attended by incommensurate phenomena. In other cases, rapid cooling may quench the disordered structure.
Many O D C structures may be viewed as a close packing of quasi- spherical, orientationally disordered groups with additional charge balanc- ing atoms present in interstices. Such simple arrangements tend to yield high symmetry, and small unit cells.
There is a large isostructural family of O D C ' s having the general for- mula A3TO4, where A is a monovalent alkali, silver or thallium atom and Τ is P, As, V, Nb, Ta or other penta valent element forming a T 04 tetrahedral group (Meyer and Hoppe, 1975). The crystal structure, as typified by N a3P 0 4 (Wiench and Jansen, 1980; Newsam, Cheetham and Tofield, 1980 a) consists of a face-centered cubic (fee) array of orientationally disordered P 04 groups with both the tetrahedrally and octahedrally coordi- nated inter-group sites filled with sodium atoms. Note that these site names refer to the number and configuration of groups surrounding each site.
The actual coordination number of atoms on these sites depends on the orientations of the neighboring groups.
This study investigates the extension of the A3TO4 structure-type to silicates, that is, finding orientational disorder of tetrahedral S1O4" groups.
An analogous silicate stoichiometry would require replacing one mono- valent cation with a divalent cation. Irvine and West (1988) make reference, without further detail, to Si-containing trisodium phosphate.
A number of silicates of appropriate stoichiometry: N a2C a S i 04, N a2M g S i 04, K2M g S i 04 and K2C a S i 04 are known (Wyckoff and Morey, 1926; Hughes, 1966) to have fee unit cells similar in size to the O D C trisodium phosphate cell. ( N a2M g S i 04 also forms an ordered wurtzite-
Crystal structure of Na2(Ca,Sr)Si04 15 like structure; Baur, Ohta and Shannon, 1981.) Metal-atom locations in Na2CaSi04 were proposed by WyckofT and Morey (1926) but neither space group nor oxygen positions were satisfactorily established. Barth and Posnjak (1932) suggested Na2CaSi04 belongs to space group Ρ2χ3 with the additional reflections allowed by this space group being too weak to detect in the powder pattern. Their proposed "stuffed cristobalite" struc- ture, with tetrahedrally coordinated Ca and Si forming a three-dimen- sionally linked network and Na atoms in network cavities, leads, however, to implausible bond lengths (e.g. Ca—O = 1.90 Â), bond-strength sums (Ca = 3.5 valence units) and calculated lattice energy (Meyer and Hoppe, 1975).
Shannon, Foris and Barkley (1980) synthesized Na2CaSi04, and several related compositions, in an ionic conductivity study. They report a negative SHG (second harmonic generation) test which strongly suggests that the space group is centric. These authors also synthesized Na2CaSi04 at 1500°C and a pressure of 30 or 58 kbar for which they report weak ad- ditional reflections interpreted as a 3 χ cubic superstructure with probable space group Fm3m. Interestingly, despite the high pressure or synthesis, the molar volume is the same as that of the one atmosphere phase.
Because of ample evidence that the accepted structure of Na2CaSi04
was wrong, and more importantly, because this compound could be an ODC silicate, a restudy of its structure was undertaken. To better determine site distribution of the divalent cation, the corresponding strontium com- pound, Na2SrSi04, as well as the 50:50 solid solution, Na2Cao.5Sro.5Si04 were also studied. Subsequent to the completion of this investigation, an unpublished study of Na2CaSi04 (Fischer, 1983), referred to in an abstract (Fischer, Tillmans, 1984) was brought to our attention and a copy of the relevant part of a thesis kindly provided us by Professor Tillmans. Our study differs somewhat in conclusions, and includes results on additional materials. Further comparison is made below.
Experimental methods
Sample preparation and characterization
Na2CaSi04 was synthesized, in a covered platinum crucible, from a mixture of CaCOj + N a2C 03 + Si02 heated in air to 1000°C for 69 h (Sample 6C). To compensate for volatilization of sodium, 10 mol% excess sodium carbonate was added. Without excess sodium, such high temperature runs yielded, in addition to the desired phase, clearly identifiable Na2Ca2Si207. A second synthesis used C a C 03 + Na2Si03 similarly heated at 900°C for 16 h, then sealed in a Pt capsule and heated for another 16 h at 1000°C, yielded Na2CaSi04 with similar X-ray powder pattern and optical proper- ties (Sample 16C). The intermediate solid solution, Na2Ca0.5Sr0.5SiO4, was

![Fig. 3. Projection of the crystal structure of (a) LiAlSi 4 Oi 0 (petalite) and (b) HAlSi 4 0 parallel [010]](https://thumb-eu.123doks.com/thumbv2/1library_info/5137070.1659854/43.629.76.555.409.675/fig-projection-crystal-structure-lialsi-petalite-halsi-parallel.webp)
![Table 4. Anisotropic thermal parameters [U(I,J) x 10**3] for non-hydrogen atoms with their e.s.d's in parentheses](https://thumb-eu.123doks.com/thumbv2/1library_info/5137070.1659854/65.629.77.558.155.527/table-anisotropic-thermal-parameters-non-hydrogen-atoms-parentheses.webp)