Role of the
143
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
Abbildung


+7
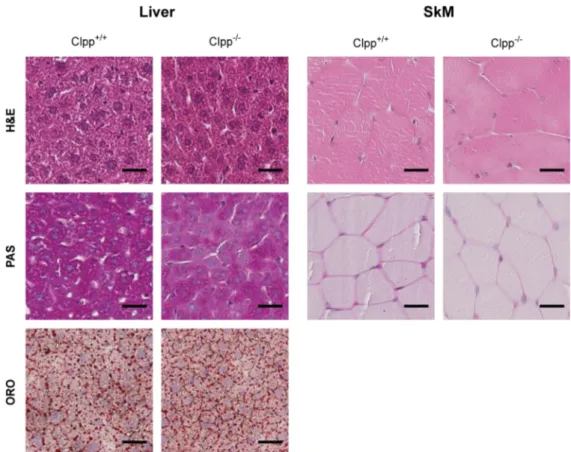

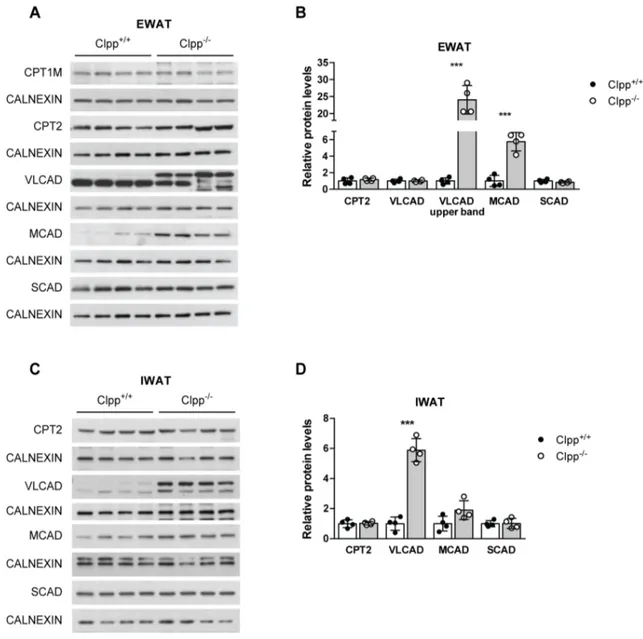
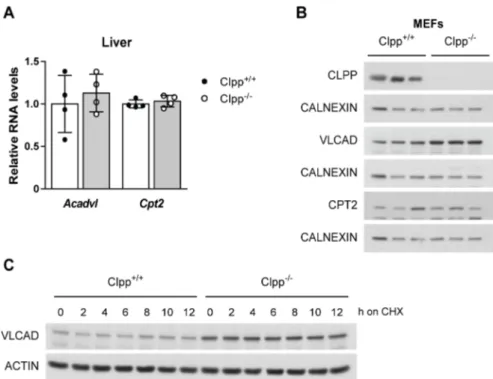
ÄHNLICHE DOKUMENTE
The main system starts from an existing emotional word dictionary (Liu et al., 2008), and use those words with strong polarities (3, the strongest) as cue for selecting sentences
Since 1981, the JEW has been organising an annual poll of projections of crude oil prices, economic growth, primary en- ergy consumption and production, the energy
The Workshop offers a unique forum for researchers working on energy perspectives to compare their sce- narios and projections on future crude oil prices, economic
the first was the recent series of political and economic upheavals which had re- sulted in sweeping changes to systems which had dominated the region since
Hence, for the pre-culture models, the number of days in ICU before blood culture extraction, the presence of catheters, fever and the presence of symptoms related to the source
In February 2011, the Greek Minis- ter for Economic Affairs, Michael Chryso- choidis, suggested that the main culprit for the economic underdevelopment of Greece is the
The main objective of the consultation was to identify the preferences of PARADIGM stakeholders with regards to core components addressing the long-term sustainability of PE.
29 Of the 617 weights attributed by Pernice to Athens the following are omitted: 1-4 early bronze pieces, 13-17 astragaloi which are not staters and have no legends, 44
