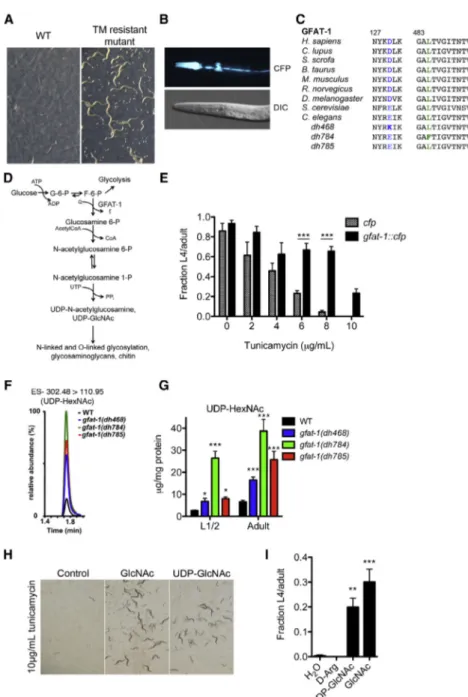
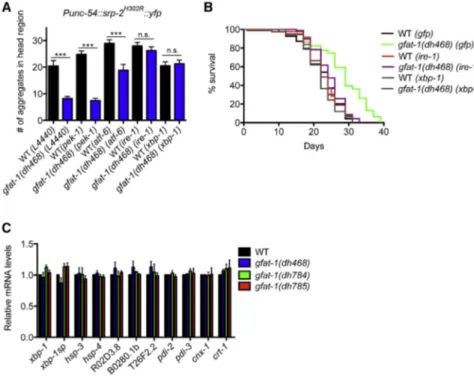
Hexosamine Pathway Metabolites Enhance Protein Quality Control and Prolong Life
Martin S. Denzel,
1,7Nadia J. Storm,
1,7Aljona Gutschmidt,
3,4Ruth Baddi,
1Yvonne Hinze,
1Ernst Jarosch,
5Thomas Sommer,
5,6Thorsten Hoppe,
3,4and Adam Antebi
1,2,3,*
1
Max Planck Institute for Biology of Ageing, Joseph-Stelzmann-Strasse 9b, Cologne 50931, Germany
2
Department of Molecular and Cellular Biology, Huffington Center on Aging, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA
3
Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), University of Cologne, Cologne 50674, Germany
4
Institute for Genetics, University of Cologne, Zu¨lpicher Strasse 47a, Cologne 50674, Germany
5
Max-Delbru¨ck-Center for Molecular Medicine, Robert-Ro¨ssle-Strasse 10, Berlin-Buch 13125, Germany
6
Humboldt-University Berlin, Institute of Biology, Invalidenstrasse 43, Berlin 10115, Germany
7