Effekte einer konsekutiven Aktivierung des MAP-Kinase-Signalwegs in humanen Melanozyten
91
0
0
Volltext
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
Abbildung
+7

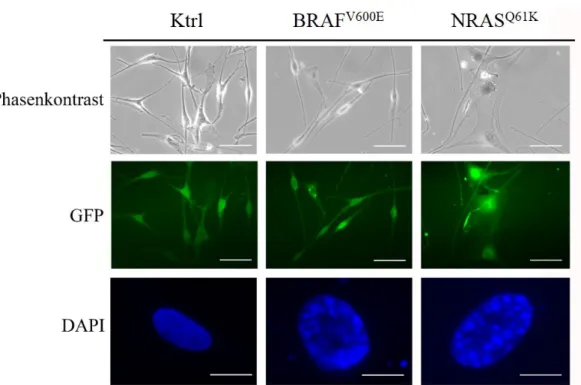
ÄHNLICHE DOKUMENTE
In der vorliegenden Arbeit konnte gezeigt werden, dass HDAC3 einen hemmenden Einfluss auf die Signalweiterleitung der MAPK11 ausübt, da die Aktivität des Transkriptionsfaktors
Anschließend unter Inkubation von Zelllinien HT 29, SW 480 und SW 620 mit Testsubstanzen Pioglitazon und Telmisartan, für maximal 72 Stunden MTT in den von uns
In allen untersuchten Melanomzelllinien konnte im Gegensatz zu Melanozyten eine stärkere Expression der IAPs (inhibitors of apoptosis proteins) sowie von cFLIP (cellular
Jedoch ist zu vermuten, dass in der TSDR des foxp3 Gens demethylierte T-Zellen eine stabile FOXP3-Expression aufweisen und es sich bei diesen Zellen um
Für den Hauptrisikofaktor bei der Pathogenese der COPD, dem Nikotin, konnte bereits eine Aktivierung der MAPK-Signalwege ERK1/2 und JNK in bronchialen Epithelzellen nachgewiesen
Der Nachweis der phosphorylierten MAPK wurde mittels Western-Blot-Analyse an Proteinextrakten der SH-SY5Y-Zelllinie durchgeführt. Als Kontrollen wurde Proteinextrakt
Zusammenfassend konnte in dieser Arbeit gezeigt werden, dass die sequenzielle Behandlung von Mikroglia mit CNI-1493 und Aβ zu einer erhöhten Ausschüttung von IL-1β
Wirkmechanismen des MAP-Kinase Inhibitors CNI-1493 auf mikrogliale BV-2 Zellen und primäre Mikroglia
Da in der vorliegenden Arbeit bereits gezeigt werden konnte, dass CNI-1493 eine Inhibition des p38 MAP Kinase Weges in Mikroglia bewirkt, wurde anschließend untersucht,
