Untersuchungen zur Signalweiterleitung in Chemo- und Photorezeptoren
141
0
0
Volltext
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(33)
(34)
(35)
(36)
(37)
(38)
(40)
Abbildung
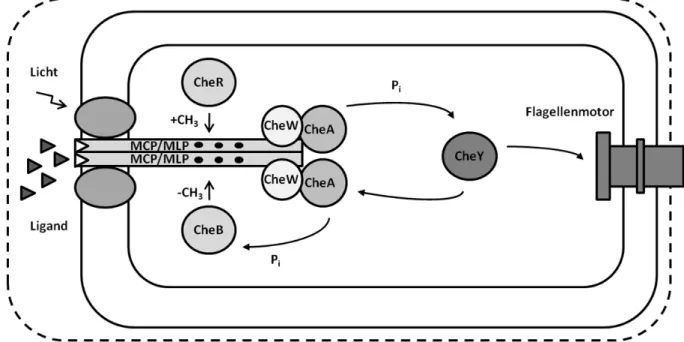
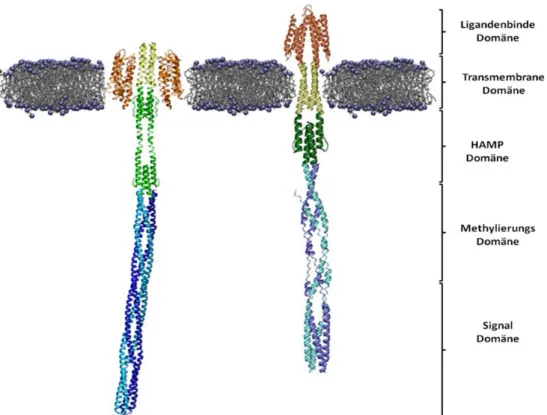
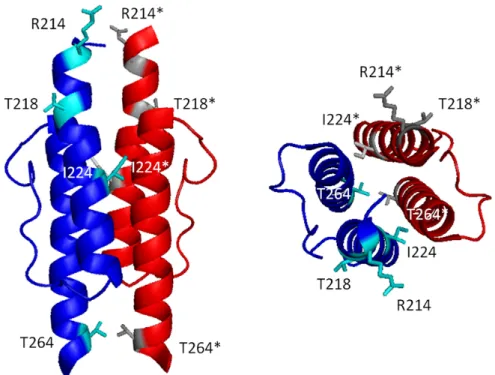
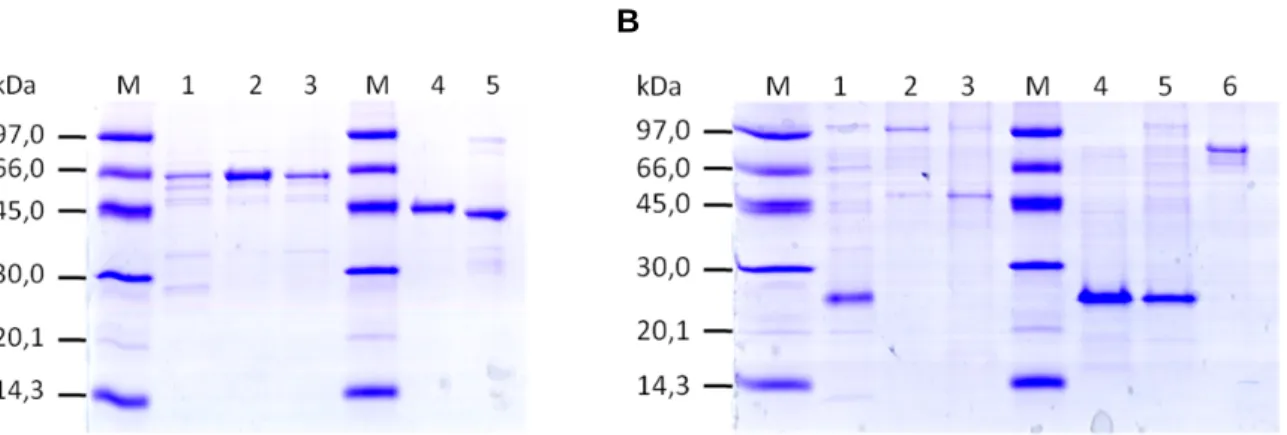
+7

![Tabelle 1: Proteinausbeuten der verwendeten Proteinkonstrukte. Protein Expressionsausbeute [mg/L Kultur] Molekulargewicht [Da] Extinktionskoeffizient [L x mol-1 x cm-1] Tar-His 1,5 61.057 39.880 (280 nm) Tar-StrepII 1,9 62.247 46.870 (280 nm)](https://thumb-eu.123doks.com/thumbv2/1library_info/3654584.1503475/57.892.99.757.139.515/tabelle-proteinausbeuten-verwendeten-proteinkonstrukte-protein-expressionsausbeute-molekulargewicht-extinktionskoeffizient.webp)
![Abbildung 16: MALDI-MS Spektrum von aufgereinigtem NpSRII-His (A) und [ 2 H, 13 C]-NpSRII-His (B)](https://thumb-eu.123doks.com/thumbv2/1library_info/3654584.1503475/58.892.111.777.111.379/abbildung-maldi-spektrum-aufgereinigtem-npsrii-his-npsrii-his.webp)
ÄHNLICHE DOKUMENTE
Business process models are typically defined using conceptual modelling languages such as BPMN or EPCs. These languages tend to be well accepted by business professionals due to
studies which concluded similar observations include Bruno and Easterly (1995), who established that a number of economies can withstand moderate inflation rates of about
Biochemical and genetic studies show that Tat and human CyclinT1 interaction requires an essential cysteine (C261 on the CyclinT1 protein) and zinc [ 60 ], indicating that
(Dieses Formular ist zur Vervielfältigung durch den Anwender dieser VDE-Anwendungsregel bestimmt.) Datenblatt einer Erzeugungsanlage – Mittelspannung.. (vom
When the SMART Interface Status Register indicates that a completion request is pending, the host processor should read the Transaction Status Register to
[r]
[r]
Da jah man auc, wie tief die Poefie Schwinds bei ihm haftet; felbft wenn er etwa feine Abenteuer mit dem Kameraden Maszfowsfi in Reihen von Bleiftiftblättern darftellt.. Er war