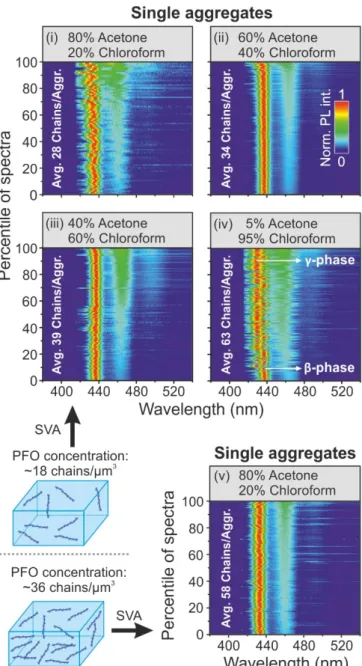
In π-conjugated polymers the connection between the elec- tronic properties and the morphology of the chain is prominent.
5
0
0
Volltext
(2)
(3)
(4)
(5)
Abbildung

ÄHNLICHE DOKUMENTE
Typical ice floes consist of flat parts, where freezing processes have increased the ice thickness continuously, and of pressure ridges that have been produced by the
The two main tasks of the expedition are the deployment of a currentmeter mooring array along a ground track of Jason altimeter satellite and the realization of a refined array
Figure 1: The price rises with demand and falls with supply under the condition of a fixed allocation of labor input and a fixed wage rate; demand is here represented by the
As for the conductivity sensor, the result of calibration shows that a set of coefficient for the conversion from the frequency to the conductivity decided at the time of the
I Über den Zeitpunkt des Empfangs kann keine Aussage getroffen werden (Unbounded indeterminancy). I Über die Reihenfolge der Empfangenen Nachrichten wird im Aktorenmodell keine
A code fragment showing how to iterate over the grid and to compute the volume of the domain by calculating the volume of each element is found at the bottom of the file
If Iran blames the United States for supporting the Syrian rebels, the US’ Arab allies argue that Washington’s failure to supply moderate Syrian rebels with
According to Alejandro Quiroz Flores, the modern state holds two comparative advantages over other forms of political organization — it is far better at directing large and
