Research Collection
Doctoral Thesis
Polymeric Friction Modifiers in Oil: Synthesis, Adsorption and Tribological Evaluation
Author(s):
Gmür, Tobias A.
Publication Date:
2021
Permanent Link:
https://doi.org/10.3929/ethz-b-000478079
Rights / License:
In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use.
Diss. ETH no. 27395
Polymeric Friction Modifiers in Oil:
Synthesis, Adsorption and Tribological Evaluation
A thesis submitted to attain the degree of
DOCTOR OF SCIENCES of ETH Z ¨ URICH
(Dr. sc. ETH Z¨urich)
presented by
Tobias Alexander Gm¨ ur
MSc ETH Materials, ETH Z¨urich Born on 19.05.1989
Citizen of Z¨urich, ZH and Quarten-Murg, SG
accepted on the recommendation of Prof. Dr. Nicholas D. Spencer Examiner HDR Dr. Juliette Cayer-Barrioz Co-Examiner Dr. Rowena Crockett Co-Examiner Prof. Dr. Lucio Isa Co-Examiner
2021
For my family
Contents
Abstract vii
Zusammenfassung ix
Acknowledgment xi
1 Introduction 1
1.1 Tribology . . . 2
1.1.1 Lubrication Regimes . . . 2
1.1.2 Friction Modifiers . . . 4
1.1.3 On Measuring Friction . . . 5
1.2 Polymers . . . 7
1.2.1 General . . . 7
1.2.2 Structure and Physics . . . 7
1.2.3 Chemistry . . . 8
1.3 Polymer Additives for Friction Modification . . . 11
1.3.1 Historical Development . . . 11
1.3.2 On the Polymers’ Chemistry and Structure . . . 11
1.3.3 PFM Working Mechanisms . . . 16
1.4 Polymers at Surfaces for Friction Modification . . . 18
1.4.1 Polymer brushes . . . 18
1.4.2 Aqueous Lubrication . . . 19
1.5 Aims of the Project . . . 22
2 Graft-Copolymers 25 2.1 Overview . . . 25
2.2 Approach One: Grafting Ratio . . . 26
2.2.1 Backbone Synthesis . . . 26
2.2.2 TMS-Trizma . . . 28
2.2.3 Post Modification . . . 28
2.2.4 Performance Assessment . . . 31
2.2.5 Conclusion . . . 31
2.3 Interlude: Hydrolysis . . . 33
2.4 Approach Two: Anchoring Units . . . 35
2.4.1 Synthetic Details . . . 35
2.4.2 Results . . . 39
2.5 Approach Three: Lubricious Units . . . 42
2.5.1 Synthetic Details . . . 42
2.5.2 Results . . . 43
2.6 Conclusion, Rationalization, and Outlook . . . 46
3 Block-Copolymers 49 3.1 Overview . . . 49
3.2 Materials & Methods . . . 51
3.2.1 Materials . . . 51
3.2.2 Method Details . . . 51
3.3 Synthesis . . . 57
3.3.1 Small Molecules . . . 57
3.3.2 Step 1: Oleophilic Block . . . 61
3.3.3 Step 2: Functional Block . . . 64
3.3.4 Step 3: Post-Polymerization Modification . . . 68
3.4 Results . . . 79
3.4.1 Resultant Architectures . . . 79
3.4.2 As a Lubricant Additive . . . 80
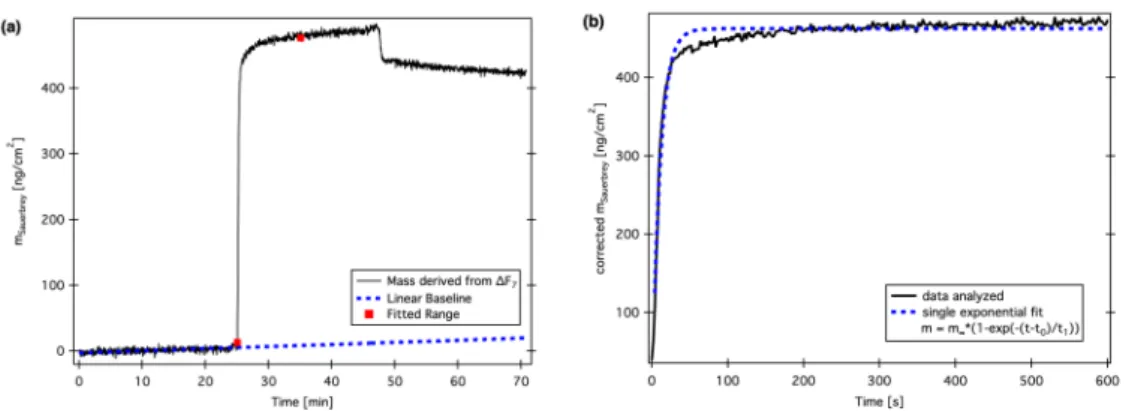
3.4.3 QCM-D . . . 80
3.4.4 MTM . . . 85
3.4.5 PoD . . . 88
3.4.6 IRIS . . . 89
3.5 Discussion . . . 98
3.6 Conclusion . . . 103
4 Conclusion and Outlook 105 4.1 Conclusion . . . 105
4.2 Outlook . . . 106
Bibliography 109
Curriculum Vitae 119
Abstract
Friction is still a thing! Whenever two surfaces are in relative motion, energy is dis- sipated, which is most of the time undesirable. Lubricants—mostly oil-based—are a common means with which to manage friction. They are composed of a base oil and a plethora of additives, friction modifiers being a key component. Innovation in the field can be directly linked to an increased efficiency and thus a reduction in CO2emission. The focus of this thesis is on polymeric friction modifiers, with novel synthesis approaches being presented, as well as their tribological characterization.
The introductory chapter lays down the foundations, focusing on the measure- ment details of friction and presenting basic chemical concepts, after which the current literature on polymeric friction modifiers is reviewed.
A first approach to develop novel friction modifiers was based on the graft- copolymer structure, where the synthesis was derived from previously developed post-polymerization modifications to create a multifunctional graft copolymer with lubricating and anchoring units. The attempts were ultimately not as fruitful as expected, which was mainly attributed to the large design space of the complex polymeric architecture.
In an attempt to simplify the general architecture, block-copolymers proved highly successful . They were synthesized and post-modified using the same chem- ical approach. One attempted structure showed superior friction reduction per- formance in rolling-sliding contacts in the mixed lubrication regime, but only in combination with catechol anchors. Other anchoring concepts or architectures did not reduce friction efficiently. Interestingly, the polymer with amine anchors did reduce wear significantly better.
Measurements with the quartz crystal microbalance indicated an adsorbed layer corresponding to a densely packed monolayer for the most lubricious polymer, while the rest adsorbed rather loosely. Finally, these results correlated with mea-
surements on a tribometer with in-situ interferometric film thickness monitoring, i.e. the friction reduction is likely due to shear-resistant films formed at the surface.
The most promising polymeric friction modifier candidate was further tested in a range of commercially available base oils, and proved to be efficient in non-polar ones, but less so in (polar) ester oils. Finally a range of additional investigations is proposed, such as the range of tribological conditions in which the additive is lowering friction, which could further our understanding of the friction-reduction mechanism.
Zusammenfassung
Mit der Reibung ist es so eine Sache. Wenn sich zwei Oberfl¨achen relativ zueinan- der bewegen, wird Energie verbraucht – meist ungewollt. Schmiermittel, meist
¨
olbasiert, sind die Werkzeuge der Wahl, um die Reibungseigenschaften zu kontrol- lieren. Sie bestehen aus einem Grund¨ol und einer Reihe von Additiven, wobei die Reibungsmodifikatoren eine Schl¨usselposition einnehmen. Innovation in diesem Bereich bewirkt schlussendlich eine Effizenzsteigerung, welche zu einem gerin- geren CO2-Ausstoss f¨uhrt. Das Thema dieser Dissertation sind Polymere als Reibungsverminderer. Neuartige Syntheseans¨atze werden pr¨asentiert und die Ad- ditive auf ihre tribologischen Eigenschaften und Wirkmechanismen hin untersucht.
Im einleitenden Kapitel werden die Grundthematiken erl¨autert. Nach einer kurzen Darlegung der chemischen und tribologischen Konzepte wird die vorhan- dene Literatur zu polymeren Reibungsmodifikatoren zusammengefasst.
Ein erster Ansatz basiert auf Co-Polymeren mit Pfropf-Struktur. Der Synthe- seweg ist abgeleitet von bereits etablierten Post-Polymerisations-Modifikations- Reaktionen, aus welchen ein multifunktionales Polymer mit verankerungs- und schmierf¨ahigen Gruppen resultiert. Die Versuche waren letztendlich nicht von Erfolg gekr¨ont, was sich haupts¨achlich auf die Komplexit¨at der Pfropf-Struktur zur¨uck f¨uhren l¨asst.
Urspr¨unglich nur als Vereinfachung des obigen Ansatzes gedacht, erwiesen sich die Block-Copolymere als ¨uberaus erfolgreich. Die etwas langwierigere Syn- these nutzte dieselbe vielseitige Post-Modifikations-Chemie. Eine dieser Polymer- Architekturen zeigte ¨uberlegene Reibungsverminderungsleistungen in Roll-Gleit- Kontakten im Misch-Reibungs-Regime, aber nur in Kombination mit Catechol- Ankergruppen. Andere Verankerungs-Konzepte oder Architekturen verminderten die Reibung zu einem geringeren Grad. Interessanterweise reduzierte ein Polymer mit Amin-Ankergruppen den Abrieb signifikant besser.
F¨ur das schmierf¨ahigste Polymer deuteten Messungen mit der Quarzkristall- Mikrowaage eine Adsorption als dicht gepackte Monolage an, w¨ahrend die ¨ubrigen Polymere weniger dicht anhafteten. Letztendlich korrelierte dies mit den Resul- taten eines Tribometers mit in-situ interferometrischer Filmdickenmessung, d.h.
es wurde gezeigt, dass die Reibungsverminderung den adsorbierten, scherresisten- ten Filmen zuzurechnen ist.
Der vielversprechendste Kandidat unter den polymeren Reibungsmodifikatoren wurde schliesslich in einigen kommerziell erh¨altlichen Grund¨olen getestet. Er er- wies sich als erfolgreich in unpolaren, nicht aber in (polaren) Ester-basierten ¨Olen.
Zum Schluss wird eine Reihe von zus¨atzlichen Experimenten vorgeschlagen. So w¨aren insbesondere Untersuchungen des Bereichs, in dem die Reibungsvermin- derer effektiv sind, von Interesse, um unser Verst¨andnis der Reibungsreduktions- Mechanismen zu vertiefen.
Acknowledgment
This thesis would not have been possible without the help and support of a few people, who I am glad to finally acknowledge here.
First and foremost I want to thank my supervisor Prof. Nicholas D. Spencer for encouraging me to pursue a PhD after my master thesis. Nic was a great role model with his constant scientific enthusiasm and optimism. Even when things were not going as planned he would always find a way to turn the mood around and help me move forward. And of course his incredibly fast editing skills have been also enormously helpful. All in all, thank you Nic for everything.
I am indebted to HDR Juliette Cayer-Barrioz for inviting me to do experi- ments with the IRIS tribometer in Lyon. The very welcoming stay, and the many insightful discussions – on tribology and otherwise - helped his thesis a lot, espe- cially in the final stages. And I would like to thank her again for agreeing to serve as a co-examiner
A big thanks goes toDr. Rowena Crockett, who kindly allowed me to use the MTM and accepted to be on my thesis committee as well. And who always had an open door for a quick coffee or insightful and funny chats, on tribology or not.
I am very grateful for Prof. Lucio Isa, who joined the committee on a short notice. He and ”the knights of the SMI” have been very accomodating for the final stages of my PhD and made the lab transition as smooth and simple as can be imagined.
I would like to thankProf. Jan Vermant for representing the Department of Materials on the comitee, his helpful comments on my research plan, and for his unobtrusive assumption of responsibility for my supervision after Nic’s emeritation.
Without my technical supervisors, I don’t think I could have achieved to cover such a broad range of topics.
First I want to thank Dr. Stefan Z¨urcher for introducing me to the post- polymerization synthesis techniques at SuSoS. His quick wit always found a new
angle on chemical issues that I hadn’t thought of.
Dr. Andrea (Andra) Arcifa, who guided me on the rough journey into tribology.
With his wisdom he paved the way for my understanding of tribological regimes and measurement techniques.
And certainly without Dr. Joydeb Mandal, his polymerization expertise, all the helpful discussions, and critical proofreading of large parts of the manuscript, this thesis would not be at the level it is now.
I would like to further thank Dr. Michael Eglin from Blaser Swisslube for supplying the base-oils and for helpful discussions.
Many thanks also go to the people who did measurements for my project: Nina Pfeffer for her patience with the initial MTM measurements. Lukas Widmer who spent 60 days in darkness for his master thesis at the AFM and was just a bit unlucky. AndTalina Graf for her countless Pin-on-Disk measurements.
Of course I owe all the people at LSST my sincere recognition as well: A special thanks goes toJosephine Baer, the cheerfully efficient heart of the group.
Rebi Huber, my former bench-neighbour, who always had an incredibly contagious positive spirit and apparently fell into the Ovomaltine-tank once. Rok Simi´c, for sharing his bed with me on conferences. Alok Goel for the smooth philosophical coffee chats. Daichi Ogawa for kindly staying heavy. Moh Divandari, Wenqing Yan, Matteo Romio andEddy Benetti the chemistry lab gang, who sometimes had to suffer my choice of music. Yvonne Gombert for the nice and candid lunch chats.
Shiva Ramakrishna, the wizard of the AFM, for his patient advice. And of course a special mention needs to go to Giovanni Cossu, who solved all the technical problems in a true McGyver fashion.
The crew at SuSoS was always extremely welcoming. A large part of the initial syntheses were conducted in their lab. My gratitude there go to: Olof Sterner, who helped immensely on the-project-that-must-not-be-named. Stefan Z¨urcher whose chemistry apprentice I was for a time. And a special thanks also toSammy Tosatti and Chris Mathis for the lively discussions. It has always been a pleasure.
Outside of work I want to thank my friends: Christophd for his LATEXexpertise and the countless coffees. And alsoJonathan andSonia for the Dunnstigskafi, the weekly anchor to look forward to and all the other distractions.
And finally my deepest gratitude goes to my family.
My mother and my brothers who made me into who I am, showed me to stand my ground and always stay gentle, and who were there for anything always.
And of course all of this would have been unthinkable without Georgina, who never stopped believing in me. Thank you for your love, the neverending motiva- tion, for enduring me at times and for heaving me out of my pits of despair. The kids are nice as well. I
IShould either of you two ever read this: Thank you for all the smiles and distraction. And the sleepless nights...
1 Introduction
”So once you do know what the question actually is, you’ll know what the answer means...”
Douglas Adams
As its title suggests, this thesis deals with the synthesis of polymers and their application as oil-based friction-reducing agents. This chapter gives first a very brief general overview of tribology and friction modifiers. After a short digression into the polymers’ properties and their synthesis, the main literature on polymeric friction modifiers (PFMs) is reviewed first from an applied perspective, i.e. litera- ture dedicated to polymers as oiliness additives. Later chapters deal with studies that emanated from works on polymer brushes, especially in the context of aqueous lubrication and the implications these had for this study.
Motivation
Tribology, a term introduced by the Jost Committee in 1966 as “The science and technology of interacting surfaces in relative motion”[1] can also be described in a simplified way as the study of friction, lubrication, and wear. Although “The Jost report” spiked quite some research and development interest[2], the topic of tribology remains a crucial issue to this day[3] . In a 2017 U.S. Department of Energy report[4] the authors estimate a prospective energy saving equivalent to roughly one fifth of the entire US energy consumption in 2016 per year by tribo- logical advancements in the U.S alone. Holmberget al.[3] estimated a prospective worldwide cost saving within 15 years of 9.7×1011 euros per year. In their “call to action” Carpicket al.[4] specify among other measures the following:
“breakthroughs in tribology [...] that are needed include: reduced viscosity lubricants[...], self-healing, ultra-thin, fluid/solid tribofilms”
1.1 Tribology
1.1.1 Lubrication Regimes
Measuring friction of a lubricated contact in a reasonable manner is not as straight- forward as one might assume, since it is a system property with many influencing factors; the exact contact geometry, mechanical properties of the solid materials, the roughness of the surfaces, the chemical nature of the surfaces, adsorbed sur- face layers, lubricant viscosity, piezoviscosity, shear-thinning, and shear-stability as well as the effective normal load and lateral speed all play a role.
When considering the textbook arguments for hydrodynamic lubrication[5]one ends up with the so-called Stribeck curve as depicted in Figure 1.1. The friction between lubricated sliding surfaces starts with high values at low speeds, shows a significant decrease with a minimum friction once hydrodynamic effects set in and solid-to-solid contact is stopped, and then increases again at higher speeds due to viscous drag. This can be grouped into three regimes that a steady state lubricated contact can be in:
• The fluid or (elasto)hydrodynamic regime, where the solid surfaces are fully separated by the lubricant due to the hydrodynamic entrainment forces. In hydrodynamic lubrication (conformal contact) the frictional forces are de- termined by the lubricant’s viscosity and the entrainment conditions. In the elastohydrodynamic regime, which occurs in non-conformal contact, the piezoviscous response of the lubricant and elastic deformation of the coun- terbodies also play a role.
• The solid or boundary regime, where the surface asperities are in direct contact with each other. The friction is only dependent on properties of the surfaces and severe wear can occur, which is generally undesirable .
• In between the two: the mixed regime, where the surfaces are partially in contact and partially supported by hydrodynamic forces.
This peculiar behaviour is named after Richard Stribeck, who as a sidenote was neither the first to describe this lubrication optimum, nor was he the one to find the universality by plotting the friction coefficient versusv·η/L(the product of entrainment speed v, viscosity η, and normal load L) the so-called G¨umbel or Hersey number[6]. Arguably he presented the most memorable graphical represen- tation.
Figure 1.1 | Stribeck Curve (1) Boundary regime (2) Mixed (3) Hydrodynamic
For practical engineering purposes, it is beneficial to be able to predict the regime a bearing is operating in for example. The film thickness in an elastohy- drodynamic contact has been approximated in the Hamrock-Dowson equation[7]:
Hmin = 3.63·U0.68·G0.49·W−0.073·
1−e−0.68∗k
(1.1) , with Hmin being the dimensionless film-thickness parameter, U, G, and W the dimensionless velocity, material, and load parameters respectively and k an ellipticity parameter. For our purposes here, the equation can be simplified into a more physically relevant form as[8]:
Hmin =C·(V ·η0)0.67·α0.53 (1.2) , with V being the mean entrainment speed,η0 the lubricant dynamic viscosity, andαthe pressure viscosity coefficient. The load, geometry, and elastic properties of the two surfaces are part of the constant C. In an EHD contact the film thick- ness evolution is generally well described by equation 1.1, lubricants with lower piezoviscosity are better described with more sophisticated models[9].
With all this in mind the general call for reduced-viscosity lubricants becomes clearer. In the (elasto)hydrodynamic regime – where most bearings are designed
to operate – the frictional losses are proportional to the viscosity, and therefore reducing it can increase efficiency. But at low speeds or high loads, the detrimental effects of solid-solid contact become more pronounced. It is in this boundary- lubrication regime is where friction-modifying compounds come into play.
1.1.2 Friction Modifiers
These compounds have a rich history and range from amphiphilic surfactant molecules (organic friction modifiers, OFM) to organo-molybdenum compounds, functionalized polymers, as well as nanoparticles[10].
Organo-molybdenum compounds, the ”gold-standard” in boundary friction re- duction, are so highly effective due to their decomposition under heat and pres- sure to form low-shear-strength molybdenum disulfide[11]. Increasingly tighter emission standards, however, call for sulfur-free lubricants in automotive indus- try. The study of polymers and nanoparticles is a rather young topic. Polymeric additives have mostly been used due to their beneficial influence on the viscosity- temperature relationship of oils[5] but can also lead to self-healing thin films as called for in the beginning of this chapter[12].
The investigation of surfactant molecules as friction modifiers dates back quite some time[13], with carboxylic acids with a relatively short (less than 20 carbon atoms) unbranched hydrocarbon tail being initially studied. The adsorption free energy of these molecules has been correlated with their frictional performance[14]. In rather recent investigations it was shown, that a single unsaturation in a C18 carbon chain in eithercis- ortrans-configuration affects adsorbed film structure[15]
and consequentially the boundary lubrication behavior[16]. Lundgren et al.[17;18]
studied the adsorption behavior of similar unsaturated fatty acids with the quartz crystal microbalance (QCM). They show that multilayer physisorption can also be encountered with highly unsaturated chains in the presence of water.
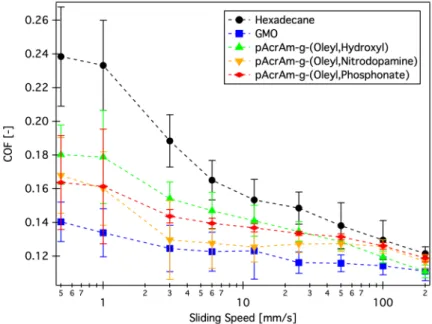
In industrial applications, carboxylic acids are not preferred, since they promote corrosion, and amines or esters are more relevant. One example commercial exam- ple being[10]glycerol monooleate (GMO), with two terminal alcohol functionalities and a C18tail with an unsaturation incis-configuration. Espinosaet al.[19;20]stud- ied GMO and novel amine and sugar-derived anchoring units and assessed their adsorption properties with QCM and correlated them with tribological perfor- mance. In their analysis a high adsorption rate constant was the relevant factor for boundary-lubrication performance. Fry et al.[21] on the other hand assessed
different OFMs related to GMO with QCM and ellipsometry and determined that a minimum film thickness is required for friction reduction to occur.
The exact friction-reducing mechanism for OFMs has not yet been entirely elucidated in all aspects[10]. On the one hand, unsaturated surfactant molecules form dense self-assembled monolayers that have a low-shear slip-plane between the opposing terminal methyl groups. The details are, as discussed above, not always coherent. To complicate things further, it is not evidenced that the monolayer shear mechanism is responsible for the effects in actual engineering contact con- ditions (with rather extreme pressures). Also soap formation by carboxylic acid OFMs, and tribochemical decomposition reactions have been reported. Recent ex- perimental and computational modelling studies[22]show a rather complex behav- ior of GMO for example, which exhibits competing adsorption and self-assembly tendencies, as well as a pressure dependent surface coverage.
1.1.3 On Measuring Friction
The aforementioned, seemingly inconsistent QCM results on adsorption need not necessarily be conflicting: neither the additives, nor the surface chemistry of the sliding partners, and most of all the exact contact conditions are not comparable.
Even when measuring in the boundary regime, a plethora of experimental factors becomes crucial.
Pin-on-Disk (PoD) measurements are most common, and involve moving a disk relative to a pin, onto which a load is applied, and the friction force being measured. The resultant pure sliding is conceptionally simple, but not always technically relevant. In most engineering systems pure sliding contact is avoided as much as possible. This is why measurement conditions of mixed rolling and slid- ing have gained popularity—especially in industrial contexts. A widely employed tool is the mini-traction machine (MTM), in which a ball and disk can be rotated independently and friction can be measured from pure sliding to pure rolling with varying loads and speeds.
Other popular laboratory tools to measure friction are the atomic force mi- croscope (AFM) and surface forces apparatus (SFA). In the former, a nanometer (up to micrometer) sized tip is used, with applied forces in the nanonewton range, while in the latter, atomically smooth macroscopic surfaces are used and the forces
are in the order of millinewtons. The translation of these measurements to effects on the macro-scale, however, is not always trivial.
As a wit has put it: “A tribometer is a good approximation for the tribological phenomena happening in a tribometer.”
1.2 Polymers
1.2.1 General
The term “polymer” commonly refers to materials consisting of large, mostly chain- like molecules. These macromolecules are formed by a polymerization process, in which several small molecules (monomers) are linked together – mostly co- valently. This terminology and the basic concept of these macromolecules were first proposed by Hermann Staudinger[23]. It is not only the chemical makeup of their monomers—or repeat units as they are termed after the polymerization process—that defines the resulting material properties, but the molecular weight distribution and structure are also crucially important.
1.2.2 Structure and Physics
The structure or architecture of polymers is only a simple straight chain in an ide- alized version. In imperfectly controlled processes there are always branch-points (as is best known for polyethylene, for example, which exists in high-density and low-density modifications due to the degree of branching). These are sometimes even intentionally aimed for and the more controlled versions of this are star- polymers, where multiple chains emanate from one central unit, comb-polymers, where multiple polymer chains are emanating from different points on a backbone chain unit, and brush-polymers, where multiple branch points have at least a tetra- functionality[24].
In copolymers (polymers consisting of different types of repeat units) the arrange- ment of the different repeat units is also crucial. A truly random arrangement is unlikely, given the fact that in any copolymerization reaction the reaction kinetics for the two monomers are rarely exactly equal, therefore the term statistical (stat) is preferred. The term block-copolymer (BCP) refers to a linear arrangement in which two chemically different polymers are attached to each other. If multiple polymers of one block are attached to a backbone polymer of another kind, similar to the comb polymers above, the arrangement is termed a graft copolymer (GCP).
The molecular-weight distribution is commonly subsumed in the two quantities Mn - the number average of the distribution - and the polydispersity index (PDI) - which is defined with the weight averageMw asP DI=Mw/Mn. Thus for a set of perfectly monodisperse chains P DI = 1, and the larger the spread, the larger the PDI. In solution, the polymers’ characteristic size, the radius of gyration Rg
scales with the length of the chain N according to
Rg ∝Nν (1.3)
, where the Flory exponentνdepends on the solvent quality (i.e. the compatibility of the polymer to the solvent) and is ∼0.6 in a good solvent[25].
1.2.3 Chemistry
Two fundamentally different polymerization methods are distinguished: In step- growth polymerization the monomers are bifunctional, and able to combine with one another randomly in both directions. Chain-growth polymerizations are usu- ally started with an initiating species, to which one monomer after another is linked. For chain-growth polymerizations there are some reaction mechanisms that offer control over the achieved molecular weight and PDI, that in their most precise form are termed living polymerization, i.e. the active chain end does not terminate[26]. There are several chemical routes to gain control in a polymeriza- tion, two of which are briefly outlined in the following.
1.2.3.1 Controlled Radical Polymerization
Figure 1.2 | (a) ATRP main equilibrium (b) RAFT main equilibrium
The two controlled radical polymerization schemes in Figure 1.2 show the main equilibria of (a) atom-transfer radical polymerization (ATRP) and (b) reversible addition-fragmentation chain-transfer (RAFT).
For ATRP[27], additionally to the monomer M, two components are needed:
an initiator mostly in the form of an alkylhalide (P X), and a catalyst consisting of a copper(I) halide Cu(I)X and a ligand L. Control is achieved in that the de- activation process is much faster than the propagation reaction, and significantly less catalyst than initiator is in the system. In this way, a chain growing from one initiator is only reacting with a few monomers upon which it is deactivated again, and another initiator unit grows with the help of the catalyst. Also there are always only a minimal amount of free radicals R• present which prevents radical combination or disproportionation reactions.
The RAFT working mechanism is different, in that first an initiator creates a free radicalP•, which is subsequently capped by the chain transfer agent (CTA), usually in the form of a thiocarbonylthio compound. From this intermediate state with a trapped radical two chains can emanate, one formed by the primary ini- tiator and one by the R unit of the CTA. This chain transfer process is the key to homogeneous growth. The Z unit of the CTA determines the solubility and reaction rate of the deactivation process towards different monomers (i.e. compat- ibility), somewhat comparable to the function of the ligand in ATRP.
The added benefit of a still-active chain end is that for both polymerization techniques there is a possibility to create block-copolymers. A detailed treatise of different approaches to create BCPs via RAFT is given in[28]. The straightforward approach is the synthesis of a homopolymer, and using that in a separate second synthesis after purification as a macro chain transfer agent (mCTA). There are certain drawbacks/caveats to be considered summarized in Figure 1.3. Aside from the end-group differences, termination reactions lead to homopolymers of the first step and tri-block copolymers, and in the second polymerization step, there will always be a homopolymer formed as well.
1.2.3.2 Post-Polymerization Modification
Modifying polymers chemically after polymerization is a technique that is older than the term polymer itself, with the vulcanization of rubber being one of the
Figure 1.3 | Polymer species formed in stepwise RAFT polymerization. Reproduced with permission[28]
first examples[29]. Post-polymerization modification is, however, generally frowned upon by traditionalist chemists, because while a modification reaction with 99 % conversion might be considered quantitative for small molecule purposes, for a post-polymerization modification the same reaction (a) might be hindered due to steric constraints and (b) results in at least 1 % of unreacted monomer units that are impossible to clean up. Nevertheless, the combination of highly efficient ”click chemistry” combined with the use of controlled radical polymerizations with a large functional group tolerance has allowed for the precise synthesis of polymers that are not accessible otherwise.
The currently most promising chemistries encompass[30]: activated esters, click reactions of the thiol-ene/yne type, nucleophilic systems based on isocyanates, etc.
In this thesis, the well-established chemistry of poly(pentafluorophenyl acrylate) (pPFPA)[31] [32] is employed.
1.3 Polymer Additives for Friction Modification
In this chapter the literature on polymeric additives for oils that have been studied for their frictional properties is reviewed. First with some historical background and then with a particular emphasis on the structure and chemical properties. The final part is dedicated to studies investigating the working mechanisms of PFMs.
1.3.1 Historical Development
Polymeric additives to oils have been largely studied for their use as viscosity modifiers (VM)[33] as well as their dispersive and detergent properties[34]. As a result, most of the existing literature on polymeric friction modifiers (PFMs) has evolved as a side product of the study of these properties. In the boundary- lubrication regime, VM-thickened oils generally shear-thin due to the high shear rates[35], which in turn leads to thinner lubricant films than expected from low- shear viscosity measurements and therefore a decreased bearing performance is observed in some cases[36]. In other instances, some VM polymers have been found to show beneficial effects on friction and wear[37;38], which cannot be explained by bulk viscosity phenomena alone. With the possibility of in situ measurements of oil-film thickness in rolling contacts in the nanometer regime, adsorbed layers of polybutadiene were measurable[39]. In a subsequent survey of commercially available VMs, the different effects polymers can have on friction and wear could be reconciled: Polymers containing polar molecular groups—polymethacrylates and polyolefins with a dispersant functionalization—showed significantly thicker films compared to non-functionalized ones[40]. These film-forming polymers did reduce friction and wear in mixed rolling/sliding as well as reciprocating conditions[41;42].
1.3.2 On the Polymers’ Chemistry and Structure
Generally used VM-polymers are poly(alkyl methacrylates) (pAMA), olefin co- polymers (OCP), poly(isobutlyenes) (PIB), and hydrogenated styrene-butadiene polymers with a molecular weight larger than 10 kDa[33] (but up to 700 kDa is usual[43]). A classical dispersant polymer is a PIB functionalized with a terminal succinimide group and a size in the range of 0.5 to 2 kDa[34]. The larger part of disclosed PFM chemistries are of the pAMA type, and will therefore be discussed first.
1.3.2.1 Acrylate PFMs
In a series of papers by Spikes et al.[44–46] in collaboration with a major additive company, the synthesis of polymers specially synthesized as friction modifiers and their detailed tribological characterization with in-situ ultra-thin optical interfer- ometry was combined for the first time.
In the first publication[44], the exact chemistry is an undisclosed pAMA with dispersant groups. The polymers are further specified to have a molecular weight between 15 kDa and 100 kDa and the structure of the polymers show a “clustered or sequenced arrangement” (i.e. a block topology is inferred). With MTM and ultra-thin optical interferometry it was shown that the structure of the polymer is more important than the chemical nature of the dispersant groups.
The second publication[45] provided more details on the chemical natures of the PFMs. Different functional units were studied (including tertiary amines, car- boxylic acid, hydroxy- and ethoxy-groups), block- and random-copolymers inves- tigated, and the effects of molecular weight addressed. The degree of functionaliza- tion (being derived from the weight fractions and molar masses of the dispersant monomers used in synthesis) was kept constant at 10 mol.%. They were blended in an API Group I mineral oil at concentrations between 8.5 and 29 wt.% to attain a constant kinetic viscosity at 120°C. The candidate PFM showing the greatest promise in the end was a poly(dimethyl aminopropyl methacrylamide-block-alkyl methacrylate) (pDMAPMA-b-pAMA) with a molecular weight larger than 50 kDa.
It showed thick films in the boundary lubrication regime and a friction coefficient that did not depend on the entrainment speed in MTM measurements.
The third installment of this investigation series[46] extended the chemistries that were studied and contained a further investigation into the effect of molecular weight and polymer concentration. The in-depth investigations were focused on morpholinylethyl methacrylate- and DMAEMA (dimethyl aminoethyl meth- acrylamide)-copolymers. The crucial effect of polymer topology—random versus block—was also reiterated. Additionally the results showed that the PFMs are continuously more effective when increasing the overall molecular weight from 23 kDa to 150 kDa, and that the lower applicability limit of concentration lies in the range of 0.3 to 1.5 wt.%. A noteworthily different behavior was observed compared to previous investigations[40]: While in the earlier investigation the film thickness decreased below the detection limit once the movement of the two surfaces was halted, for these novel polymers a residual separation of several nanometers was measured, indicating a strongly adsorbed film. In more severe pin-on-disk condi-
tions of the high-frequency reciprocating rig (HFRR) the PFMs were less effective.
Similar PFMs as above were studied in two publications by Masuko et al.[47;48]
with the raw polymers supplied by the same additive company.
In the first study[47] different pAMA-stat-pDMAEMA PFMs were studied in a block-on-ring macrotribological test, the surfaces being analyzed after sonication in hexane with TOF-SIMS and AFM adhesion and friction studies. The PFMs contained up to 0.6 mass.% of nitrogen, which corresponds to a degree of func- tional monomers of 10 mol% (assuming the alkyl to be a dodecyl chain), and had molecular weights of 7.5 or 20 kDa. They were studied in a polyalphaolefin oil at a fixed concentration of 10 wt.%. The macrotribology showed a decrease in friction in the boundary-lubrication regime, most notably with the larger PFMs with the higher degree of functionalization. From ToF-SIMS the presence of an absorbed boundary film / tribolayer derived from the PFMs was evident. These adsorbed boundary films were ex-situ still measurably different in AFM friction and adhesion properties.
The more recent study[48]concerned itself with the comparison of PFMs and OFMs with tertiary amine (i.e. DMAEMA) and alcohol functionalizations. The different behaviors of OFMs (small molecules with a single adsorption site) versus PFMs (large molecules with multiple adsorptive units) were especially evident in mea- surements of the friction coefficient with a continuously increasing temperature.
While for the OFMs and the alcohol-PFMs a critical desorption temperature was found—beyond which the coefficient of friction went up significantly—their amine- modified PFM counterparts showed none of these effects in the investigated tem- perature range. Additionally the effect of solvent polarity was investigated, and an increase in transition temperature was found with increasing dielectric constant.
Cosimbescu et al. presented[49–51] the complete synthesis of polymers that were subsequently characterized for their VM and PFM performance. In[49], a free-radical polymerization of methacrylates with similar functionalities as in[45]
is described. The resulting “low molecular weight” polymers had sizes below 60 kDa and showed promising effects for use as VMs at a concentration of 12.5 wt.%
in a Group III mineral oil. The friction reduction and wear prevention could be measured, and was comparable to what had been noted earlier[45].
In[50] the RAFT synthesis of random copolymers of alkylmethacrylates with a range of polar monomers is presented. The most promising candidate for friction and wear reduction was a poly-(dodecyl methacrylate-stat-vinylimidazole) with a molecular weight of 45 kDa at a concentration of 2 wt.% in “commonly used hy-
draulic fluid base oils”.
Another approach to novel PFMs was taken in[51], where the authors synthe- sized an array of statistical copolymers with the non-polar unit being a dodecyl methacrylate, and the polar, surface-active part being a methacrylate containing different types of ionic liquids. Their molecular weights spanned the range from 9.5 to 207 kDa and were produced with a free-radical and RAFT polymerization methods. The friction and wear tests conducted at 5 wt.% in a polyalphaolefin showed a great wear reduction potential, while the effect on friction was negligible.
Finally, in[52]the film-thickness evolution in pure rolling, the traction behavior with a fixed slide-to-roll-ratio of 50 %, and the boundary friction in pure sliding contact were compared. The polymers investigated were three different unfunc- tionalized pAMAs with some degree of very short-chained alkyl methacrylates, that induced a significant degree of polarity which in turn allowed for some sur- face adsorption. The results of the pure rolling were similar as before. At high speeds significant shear thinning was measured, while at low speeds an adsorbed polymer layer was visible. The traction measurements were not what one might expect from the film-thickness results: the polymer showing the thickest adsorbed film showed the higher friction, while the practically non-adsorbing one decreased friction significantly. Thus it seems that thick films are not a guarantee for opti- mal friction performance. In pure sliding boundary friction, however, the polymer showing the thickest film did exhibit the lowest coefficient of friction.
1.3.2.2 Other Monomer Chemistries
The literature on non-PAMA based PFMs is a bit sparser until now, but worth mentioning nonetheless.
Guegan et al.[53] focused on the interplay of different types of friction mod- ifiers. The combinations of two undisclosed “poly” additives with and without OFMs and molybdenum-based FMs were investigated in three different tribolog- ical tests – MTM, HFRR, and unidirectional PoD. The results were dependent on test conditions, i.e. under some measurement conditions two additives could behave antagonistically, while in another test they were synergistic.
In a paper by Zhang et al.[54] the tested PFMs were produced by modifying a commercially available PIB with an ionic liquid terminal group. The produced
compound was shown to increase friction and decrease wear when used on its own.
In combination with ZDDP (the most common anti-wear additive), friction coef- ficient and wear could both be significantly reduced.
Van Ravensteijn et al. have presented[55] a work on organic-inorganic hy- brid star polymers as PFMs and VMs. Their approach was to grow statistical copolymers of methylmethacrylate and octadecyl methacrylate from organic and inorganic core segments with the number of initiating sites varying from 1 to 9.
The lubrication performance was assessed in a high-speed surface forces apparatus (i.e. at pressures significantly lower compared to the studies mentioned up to now) and complementary film thickness measurements with quartz crystal microbalance (QCM) were conducted. It was found that the star polymers with a higher degree of branching resulted in thicker adsorbed films and better frictional performance.
A rather recent report by Murdoch et al.[56] discussed the use of OCP func- tionalized with a low amount of nitrobenzene moieties through succinic anhydride linkage. These FOCP (originally designed for their unique temperature-viscosity effect) have shown to significantly reduce friction in the boundary regime as mea- sured in an AFM setup. Accompanying normal force versus distance and QCM measurements indicate that the adsorbed FOCP films do induce a steric repulsion of the surfaces, but also that single chains can “bridge” the contact and lead to high adhesion and friction. The best frictional performance was achieved with an adsorbed FOCP layer and a lubricant liquid only containing OCP.
1.3.2.3 Patent literature
The patent literature is in its nature a bit less transparent. The patent most closely related to[44–46] is[57], which specifies the chemistry further. Primarily a block-copolymer synthesis via a one-pot RAFT polymerization is claimed.
In[58;59] the synthesis of polymers for use as PFMs is claimed. They are produced in three steps: after a free-radical copolymerization of different alkyl- methacrylates and styrene, a subsequent modification with maleic anhydride and a grafting of N,N-dimethylaminopropylamine yielded a similar anchoring unit as in[46].
In patents[60;61]statistical copolymers of different alkylmethacrylates (or acrylates, the authors are a bit fuzzy on that detail) and hydroxyethylmethacrylate – a polar monomer described in[46] as well – are claimed. The main difference of the two
patents (filed by different companies notably) is in molecular weights of the PFMs;
while in[60] the examples have a Mn of 3 kDa, in[61] the preferred embodiments were in the range of 25 kDa.
Croda[62] has patented an approach to synthesize PFMs by linking PIB and poly(ethylene glycol) to yield star-like copolymers with two hydrophilic and two hydrophobic blocks. A similar PFM is patented by Huntsman[63], in which a polyisobutylene-block-poly(ethylene glycol-stat-propylene glycol) is described by linking the two blocks with a succinic anhydride-amine chemistry. Another block- copolymer approach to PFMs is patented by BASF[64]. Here a triblock copoly- mer consisting of a central poly(tetrahydrofuran) block that is flanked by two poly(butylenoxide) units is claimed.
1.3.3 PFM Working Mechanisms
As expanded on before, a differentiation between the different lubrication regimes is crucial. For polymeric additives in general mostly the EHD regime is considered, where film thickness and friction are the two main parameters of interest. They also happen to be generally not directly related[8].
For the studies on PFMs, their contribution on film thickness is more in focus.
The results of the first set of measured film thicknesses[39–42] was subsequently compared to a layered viscosity model[65], where a similar algorithm leading to equation 1.1 was used with assuming a layer atop each sliding surface with vary- ing thicknesses and increased viscosity. This model was motivated by the absence of a measurable film once the motion was halted. An agreement with the measured results of one investigated VM was reached with a modelled film thickness of 18 nm and a surface viscosity of 4 to 16 times that of the bulk liquid.
A more recent investigation on similar polymer additives (mainly in use for their VM properties) is presented in[66]. Here the focus was on the independent mea- surement of shear thinning, and the application of these results to an accurate film thickness prediction. This worked well for film thicknesses significantly larger than the radius of gyration of the polymers (40 nm in this case). In the very thin film regime a similar adsorbed film with a thickness of one or two radii of gyration (depending on the polymer chemistry) was evidenced in accordance with the above.
The friction of an EHD contact is in generalcomposed of the sliding (Couette)
and the rolling (Poiseuille) contributions, whereby the latter is only relevant at very low slide-to-roll ratios and therefore mostly neglected. The prediction of the sliding friction from bulk lubricant properties is complicated by the fact that at the high pressures and shear rates the lubricant is far from the Newtonian idealization.
In practice frictional measurements of an EHD contact are fitted preferably[8] with a shear thinning model of the Ree-Eyring type[67].
A more sophisticated approach is presented in[68], where the friction and thick- ness response of industry supplied PFM solutions (with undisclosed chemistry) were interpreted with a layered Eyring-type model. Notable conclusions were that the surface layer was slightly thicker under shear than in pure rolling, and that the base oil is crucial for the PFM behavior – a fact that is mostly neglected in the previously discussed literature.
1.4 Polymers at Surfaces for Friction Modification
1.4.1 Polymer brushes
Polymer brushes[69], i.e. polymers attached to a solid surface, remain a topic of interest for surface science not only for biological applications[70], but also because it has been demonstrated that they show excellent lubrication properties[71;72].
The explanation for these excellent lubricating properties – measurements of coefficient-of-friction<0.001 are reported – is currently based on entropy effects[71]. With a high grafting density in a good solvent, the polymer chain needs to stretch away from the surface, taking it far away from its thermodynamically optimal, coil-like configuration. When the polymer chains are attached to a surface at sufficiently high coverageσ > σc, their characteristic size, the height scales with
h∝N·σ1/3
, whereσc ∝N6/7 [73]. Compared to equation 1.3 the striking difference is that the height of the brush scales linearly with the length of the chain, which implies that it is in a strongly stretched configuration. The regime where this height relation holds is usually referred to the polymer-brush regime, whereas at lower coverage (where the crowding is not sufficient to force the polymer chains to stand up) the polymers resemble a more mushroom-like structure and the characteristic height behaves sublinearly.
The scaling relations given above are the result of a free-energy-balance argu- ment. With a more detailed treatment of the chain conformations as well, it was found, that (while the height linearity still holds) the monomer density profile from the surface follows a parabolic form[69]. For the lubricity behavior between two polymer brush coated surfaces, this means that there is a layer of solvent present in between the brushes only due to their equilibrium conformation. This allows for fluid-film sliding similar to that in the hydrodynamic regime—even at negligible velocities. Another factor that is responsible for the excellent lubricity is the os- motic pressure of the trapped solvent, which maintains a fluid film in between the sliding surfaces. However, these impressive properties only appear at pressures in the lower MPa range[72]. At increasing pressures two polymer brushes will start to interdigitate, and the lubrication mechanism changes[74]. This “critical interdigi- tation pressure” has also been shown to be significantly affected by the topology of the brushes[75].
The two distinctly different approaches with which polymer brushes can be produced are commonly called grafting-from and grafting-to[72]. Grafting-from is realized by polymerizing a monomer from an initiator molecule permanently bound to a surface – growing the chain from the surface so to speak – whereas in the grafting-to approach the polymer is in solution with a reactive chain end that sticks to the surface. There are several pros and cons of the two approaches:
Grafting-from results in brushes with a high grafting density but lacks the pre- cise control (and measurement possibility except for very select cases[76]) of the polymer dispersity. It also requires a rather high-level of synthetic chemistry, and is impractical for industrial applications. Grafting-to on the other hand has less challenging chemistry, but often suffers from the low grafting densities that can be usually achieved. It needs to be stressed that most of the theoretical treatments above are assuming a stretched brush, i.e. that the linear height to chain length relationship holds. As this applies only to either very long chains or a high grafting density, this need not be applicable to grafted-to polymer brushes. The goal of self-healing, however, strongly favors the usage of a grafting-to approach, making it vastly more interesting from an application point of view.
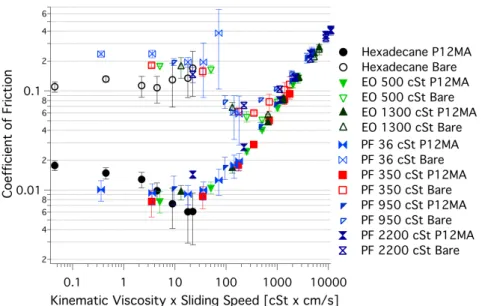
In his PhD thesis Bielecki studied the effects of polymer brushes on oil-based lubrication[77]. In Figure 1.4 the result of a Stribeck-like study is shown. For this a 250 nm poly( dodecyl methacrylate) coated silicon-dioxide tribopair is compared to the equivalent bare surfaces in different oils. It is clear that the onset of the boundary regime is significantly shifted to lower speeds, and the coefficient-of- friction stays below 0.02 for low speeds. However, the grafting-from methodology limits the application of these findings.
1.4.2 Aqueous Lubrication
In water-based lubrication the application of polymer brushes is somewhat more advanced. For example in[12] a polymer-brush-based improvement of coefficient of friction with a grafting-to approach that shows self-healing properties in an aqueous system. The lubricating molecule here is a graft copolymer with a poly- L-lysine (PLL) backbone onto which poly(ethylene glycol) (PEG) sidechains are grafted. The positive charge of the PLL makes it adhere to the negatively charged silicon dioxide, while the hydrophilic PEG-chains form a brushy layer that works as a steric barrier. It was also shown that the lubricating effect of PLL-g-PEG is only temporary in the first cycle when the surface is only incubated in PLL-g-
Figure 1.4 | Stribeck-like plots for bare–bare SiO2-borosilicate glass ball (empty mark- ers) and for double-sided 250 nm (dry thickness) grafting-from P12MA brush– brush functionalised tribological contacts (filled markers), with different base fluids. Reproduced with permission[78] .
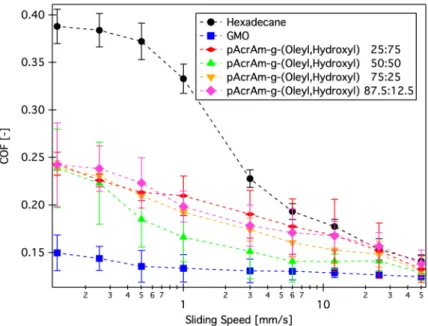
PEG solution and tested without it. The lubricity can be regained upon injection into the bulk solution. The self-healing effect was further evidenced as depicted in Figure 1.5, where a glass disk was incubated in a green fluorescently labelled PLL-g-PEG, and rubbed against a steel pin in a solution of red labelled PLL- g-PEG. However, the resulting coefficient-of-friction is with 0.15 not competitive with standard oil formulations.
The original intention of PLL-g-PEG in water as presented above was col- loidal stabilization[79], whereas it has been later shown to have excellent protein- resistance properties[80]. Although lubricity and protein-resistance of polymer brushes do not need to correlate, in the case of PLL-g-PEG they seem to do[81].
1.4.2.1 On PLL-g-PEG architecture
A detailed study of the structure-property relationship was undertaken for the anti-fouling properties by synthesizing a large set of polymers with different ar- chitectures. Pasche[82] synthesized an array of PLL-g-PEG variants with different grafting ratios and respective sizes. Her results implied that there are different reasons for an adsorbed film to be not successfully working in turning surfaces
Figure 1.5 | Fluorescence microscopy image of the disk following pin- on-disk tribometry experiment with a steel/glass tribopair . Reproduced from[12]with permission.
protein resistant. These rationalizations are sketched in Figure 1.6. The grafting ratio can be (a) too low for an sufficiently dense film to be formed or (b) too high, such that the ordered adsorption on the surface is hindered by steric effects. A too-high overall molecular weight can result in disorganized adsorbed layers (c), which do not repell proteins efficiently. Additionally the optimal grafting ratio is dependent on the molecular weight of the side chain.
Figure 1.6 | Influence of polymer architecture on the conformation of the formed adlayer and the subsequent implications for protein resistance. A high grafting density is needed to acheive protein resistance (d), while a too-low brush density (a) results in non-resistant coatings. A too-high grafting ratio on the polymer backbone (b) results in stiff polymers that adsorb in tail-on conformations. Too-high molecular weight (c) can induce looped conformations. Reproduced from[82]
In a later study the lubricious performance of PLL-g-PEG systems has been shown to correlate with the ratio L/(2·Rg) where L ≈ σ0.5 corresponds to the average chain spacing[83].
1.5 Aims of the Project
The original idea of this project was to synthesize a PFM that works in oils with a multifunctional comb structure – inspired by the works on PLL-g-PEG – which would be a novel architecture approach in the field of PFMs. The synthetic approach chosen was based on a successfully used post-polymerization modifica- tion[32], that allows different chemistries and grafting structures to be efficiently tested while decoupling the effect of polymer backbone structure completely.
In a second approach, block copolymers were investigated as a simplifica- tion of the first attempts with a straightforward RAFT synthesis approach. The same post-polymerization modification chemistry was used to study the effect of adsorbing-unit chemistry, while the effect of molecular weights of the blocks were studied independently. This block architecture compares also to established struc- tures[46], but for the influence of relative block sizes no research efforts are pub- lished.
The envisioned structures in solution as well as in the adsorbed state are de- picted in Figure 1.7. The merit for the GCP approach (Figure 1.7 (1a) and (1b)) is the increased (maximal) surface grafting density of lubricious chains, which is the prerequisite for a polymer brush. The “pre-grafting” of the lubricious chains onto the backbone will also result in a predetermined interchain distance, and higher surface grafting densities should be accessible compared to monofunctionalized chains, since the relative “footprint” of a single chain extending into solution will be significantly smaller compared to block copolymers. The BCP-PFM (Figure 1.7 (2a) and (2b)) on the other hand requires less conformational rearrangement to adsorb to a surface and the design space is less complex.
Figure 1.7 | (1) graft copolymer (2) block copolymer (a) in solution and (b) adsorbed onto a surface
2 Graft-Copolymers
”A goal is not always meant to be reached, it often serves simply as something to aim at.”
Bruce Lee
2.1 Overview
This chapter deals with the development of graft-copolymer based friction mod- ifiers. It was ultimately not as successful as the block approach presented in chapter 3. The presentation here therefore does not follow the traditional scien- tific differentiation between methods, results, and interpretation, but is rather in a chronological order emphasizing the learnings and gradual increase in complexity.
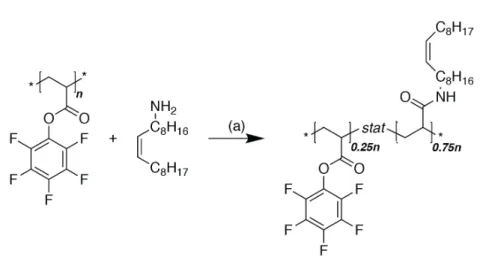
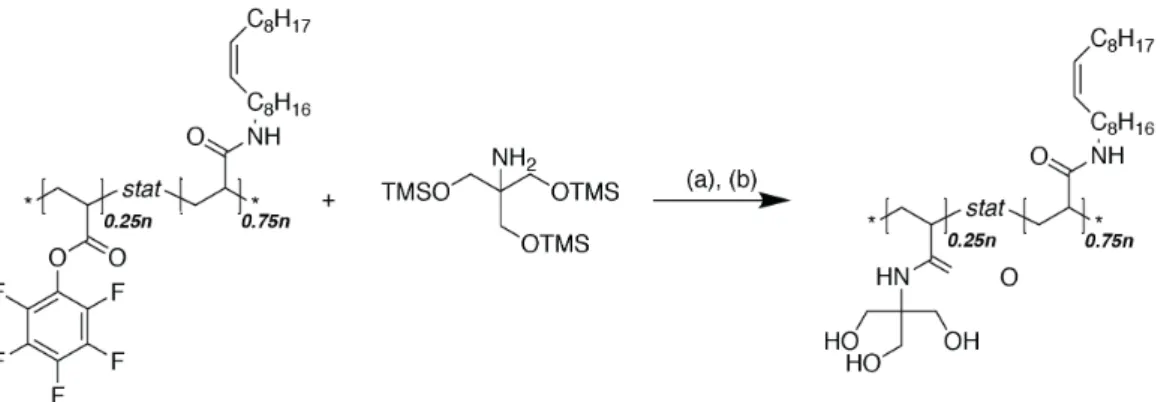
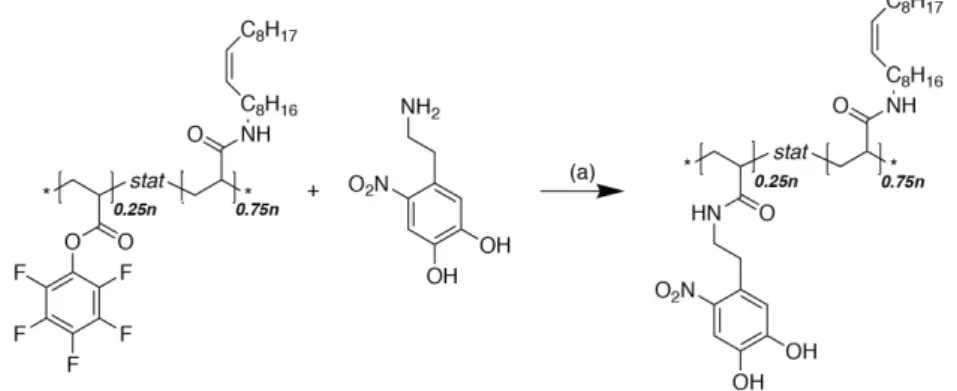
The aim was to create a structure similar to what has been proven to be successful in the PLL-g-PEG system for aqueous lubrication[84] (see also chapter 1.4.2), and transfer the knowledge to produce graft-copolymeric friction modifiers that function in oil, i.e. non-polar liquids. The synthetic procedure was based on the multimodal binding approach presented by Serrano et al.[85] outlined in Figure 2.1. In a first step a functional polymer is produced with a defined molecular weight. The polymer is then partially modified with an oleophilic lubricious group, after which the surface active anchoring units is added.
Figure 2.1 | Schematic summary of the graft-copolymer synthesis. (1) Functional back- bone polymerization (2) Modification with lubricious units (3) Modification with anchoring units
The investigations presented here discusses the effect of the grafting ratio, different anchoring, and lubricious units. The hurdles that were encountered in synthesis and characterization are emphasized. In a final section the implications for the feasibility of this approach are discussed.
2.2 Approach One: Grafting Ratio 2.2.1 Backbone Synthesis
Figure 2.2 | RAFT scheme for the poly(pentafluorophenyl acrylate) synthesis. (a) Toluene, 80°C
The precursor polymer poly(pentafluorophenyl acrylate) (pPFPA) was synthe- sized according to a previously described RAFT polymerization recipe[85]. The monomer pentafluorophenyl acrylate was supplied by SuSoS and freshly distilled.
77 g of the monomer was added to a 250 ml two-neck round-bottom flask, together with 774 mg chain transfer agent (2-(Dodecyl thiocarbonothioyl thio)propionic acid, Aldrich), and 129 mg initiator AIBN (azobisisobutyronitrile, Aldrich, re- crystallized from EtOH) and 77 ml toluene. The solution was degassed by three freeze-pump-thaw cycles and the polymerization started at 80°C. The reaction was run overnight, after which the purification was acheived by precipitation in MeOH three times. After drying at high vacuum a slightly yellow powder was obtained. Yield: 64 g (82 %)
The1H-NMR and19F-NMR spectra in CDCl3are presented in Figure 2.3. The spectrum in (i) is reasonably close (e.g. indistinguishable) to that in Figure 4.2 in[85], where a rigorous peak assignment has been done. The three peaks in (ii) atδ =−153.2 ppm,δ =−156.8 ppm, andδ =−162.2 ppm can be assigned to the ortho-, para-, and meta-positions, respectively, of the fluorine with respect to the oxygen.
Figure 2.3 | Regions of interest in (i)1H-NMR (ii)19F-NMR for the precursor polymer poly(pentafluorophenyl acrylate) in CDCl3
2.2.2 TMS-Trizma
Figure 2.4 | Scheme for the TMS-Trizma synthesis. (a) DMF, 125°C
1 g of trizma (Sigma-Aldrich) was added to a two neck round bottom flask under nitrogen. 8 ml of each DMF (VWR) and hexamethyldisilazane (HMDS, Sigma-Aldrich) were added and the reaction run at 125°C overnight. The resultant product was reduced under high vacuum, filtered through a short silica gel column with ethyl acetate and reduced again to yield a clear yellow liquid (2.5 g).
1H-NMR (CDCl3, 300 MHz): δ= 3.32 ppm (C CH2 O),δ = 0 ppm (TMS) with an area ratio of 2 : 8.9.
2.2.3 Post Modification
Figure 2.5 | Scheme for the polymer postmodification. (a) TEA/THF, 70°C, (b) THF, 70°C, (c) DCM, (MeOH/H2SO4)
The coupling reaction was performed with the goal of obtaining four different ratios: 87.5, 75, 50, and 25 % of oleylamine. The reactions consistently followed the same recipe: for 1 g of pPFPA, the desired molarity of oleylamine (Acros Organics) - i.e. grafting ratio times mol of PFPA units - was added together with 10 ml of tetrahydrofuran (THF, VWR) and 1.7 ml triethyl amine (TEA, Merck) to a two-neck round-bottom flask with a reflux condenser. The reaction was kept at 70°C overnight. The TMS-trizma was added afterwards in a 50 % excess. After another overnight reaction the product was cleaned by precipitation in MeOH.
The protection group was removed by re-dissolution in 10 ml DCM and stirring overnight in the presence of 1 ml (MeOH/H2SO4) mixture (100/1). The polymer was then precipitated in MeOH and dried under high vacuum. Yields for 87.5, 75, and 25 % oleylamine functionalization: 87.6 %, 83 %, and 10.8 %.
The resulting NMR spectra (CDCl3, 300 MHz) are shown in Figure 2.6 with the findings briefly outlined here. Firstly, the spectrum of the variant with nominally 25 % oleylamine showed remaining contaminants, which is due to the inadequate purification procedure that also resulted in a diminished yield. The olefinic peak for the oleyl group can be clearly observed at δ= 5.35 ppm, the expected peak of the C CH2 O units of the trizma moieties at approximately δ = 3.32 ppm is a bit less evident. In an effort at quantification, the entire range from 3 to 4.75 ppm was therefore integrated and the results listed in Table 2.1.
Remaining 1H-NMR peaks assignment: δ = 2.02 ppm (CH C O), δ = 1.28 ppm (C CH2 C),δ= 0.90 ppm ( CH3)
Figure 2.6 | NMR spectra of the four variants with different grafting ratios of oleylamine and trizma. From top to bottom: 25, 50, 75, and 87.5 % of nominal oleyl content
Table 2.1 | Integral intensity of the trizma peaks in the1H-NMR spectra shown in Figure 2.6, with the oleyl-peak normed to 1. (a) assuming full conversion of the first grafting and no side reactions. (b) measuring the area in the range 3 ppm to 4.75 ppm
% oleyl theory (a) measured (b)
25 9 5.36
50 3 2.97
75 1 1.04
87.5 0.43 0.94
2.2.4 Performance Assessment
The polymers were dissolved in hexadecane at 0.5 wt %. The variant with 75 % showed limited solubility, i.e. the solution was clearly opaque as opposed to the others. It was analyzed nonetheless. The four investigated polymers were com- pared to lubricants containing GMO (glycerol monooleate) and the pure base oil hexadecane.
Pin-on-Disk measurements were performed on a Bruker UMT2 instrument at room temperature. The disks (100Cr6) were freshly polished to a roughness
<20 nm. Ball (6 mm diameter, 100Cr6) and the disk were sonicated in toluene and isopropanol before use, the setup assembled and shimmed. At first three repeat Stribeck-type measurements were performed with decreasing speed ramps from 500 mm/s to 0.125 mm/s with pure hexadecane only. The normal load was 5 N which resulted in a Hertzian maximum contact pressure of 1.1 GPa. Subse- quently, on a new track on the disk, three repeat Stribeck steps were done with the lubricant solutions. For every step at least 3 full revolutions were performed and only the last half of measured friction coefficient were considered, to avoid transient effects.
The results presented in Figure 2.7 are averages and 95 % confidence intervals of the three repeat measurements. The conclusion of these results was that on the one hand, the PFMs seemed to reduce friction to some extent. On the other hand (and more relevant) no effect of grafting ratio was observed.
2.2.5 Conclusion
The trends of measured grafting ratios shown in Figure 2.1 do follow the expected trends, but the results hardly suggest a precise control over structure. This is not entirely unexpected since post-synthesis NMR quantification was also not success- ful in earlier studies[85].
The results indicate that neither was the synthesis very controlled in grafting ra- tio, nor did the resulting polymers show significant benefits as friction modifiers compared to the GMO standard. Additionally, polymers with different grafting ratios did not show any trend in the friction measurements that could warrant further conclusions.
Figure 2.7 | Pin on disk measurement results for the grafting ratio modification ap- proach.
2.3 Interlude: Hydrolysis
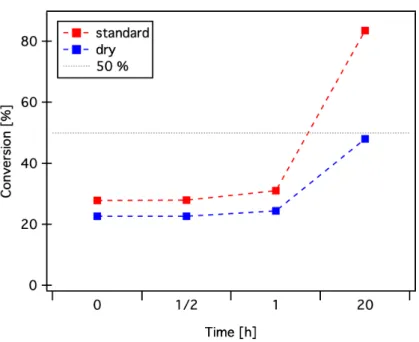
While searching for an explanation for the encountered characterization issues, the topic of hydrolysis emerged. Although the pPFPA-ester chemistry is claimed to have higher hydrolytic stability than poly(N-hydroxy succinimid)-esters[30], we could not find any explicit quantification published. For this reason a brief study of the impact of moisture on the pPFPA modification was carried out. Two mod- ification reactions were started simultaneously with oleylamine in a 50 % molar ratio (referring to the PFP-units). The first one as described in chapter 2.2.3 with standard reagents, while for the second one the reaction vessel was evacuated and refilled with nitrogen, dry THF was transferred via syringe and oleylamine and TEA were added under nitrogen. Figure 2.8 shows the results of aliquots subjected to19F-NMR analysis (Acetone D6, 300 MHz) after different times at 50°C. The conversion was calculated by the ratio of the broad pPFPA peaks atδ =−154 ppm, δ =−157 ppm, andδ =−164 ppm versus the sharp peaks of pentafluorophenol at δ =−170 ppm,δ=−172 ppm, and δ= −189 ppm.
The results’ implications are quite straightforward: Dry conditions are necessary.
Otherwise the effectively available functional units on the backbone for a further modification are not controllable. Also the architectural quantification via this route needs to be taken with a grain of salt, since the results are only a quantifi- cation of the PFP liberation, and not strictly of the amidification process.
Figure 2.8 | Conversion evolution of a reference pPFPA modification reaction aiming at a final functionality of 50 %.