Untersuchungen an neuartigen Serin-Threonin-Kinase-Inhibitoren für die In-vivo-Visualisierung von Signaltransduktionswegen des quergestreiften Muskels
185
0
0
Volltext
(2)
(3)
(4)(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
Abbildung
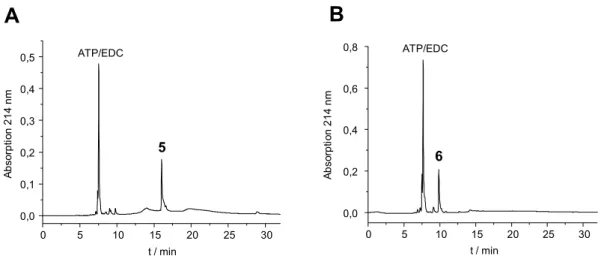
![Abb. 16: HPLC-Chromatogramme und Massenspektren von (A) P 3 -(N ε -(N-FITC-[K5, V6, A7]-CONH 2 - -Kemptide)-lysyl)-P 1 -adenosin-triphosphat 2 und (B) P 3 -(N ε -(N-TRITC-[K5, V6, A7]-CONH 2 -Kemptide)-lysyl)-P 1 -adenosin-triphosphat 3 nach Reinigung](https://thumb-eu.123doks.com/thumbv2/1library_info/3651450.1503322/43.892.137.784.292.828/chromatogramme-massenspektren-kemptide-adenosin-triphosphat-kemptide-triphosphat-reinigung.webp)
![Abb. 17: Autoradiogramme der Verdrängung von [γ- 32 P]-ATP durch ATP und P 3 -(N ε -(N-acyl-O-methyl)-lysyl)- -(N-acyl-O-methyl)-lysyl)-P 1 -adenosin-triphosphat 1 bei der Phosphorylierung von c-C1C2 durch PKA-C](https://thumb-eu.123doks.com/thumbv2/1library_info/3651450.1503322/45.892.275.605.397.713/autoradiogramme-verdrängung-methyl-lysyl-methyl-adenosin-triphosphat-phosphorylierung.webp)
+7
![Abb. 18: Autoradiogramme der Verdrängung von [γ- 32 P]-ATP durch [V6, A7]-Kemptide und N-Fmoc-[K5, V6, A7]-CONH 2 -Kemptide 4 bei der Phosphorylierung von c-C1C2 durch PKA-C](https://thumb-eu.123doks.com/thumbv2/1library_info/3651450.1503322/46.892.247.635.193.526/abb-autoradiogramme-verdrängung-kemptide-fmoc-conh-kemptide-phosphorylierung.webp)
![Abb. 19: Autoradiogramme der Verdrängung von [γ- 32 P]-ATP durch ATP und P 3 -(N ε -(N-FITC-[K5, V6, A7]- A7]-CONH 2 -Kemptide)-lysyl)-P 1 -adenosin-triphosphat 2 bei der Phosphorylierung von c-C1C2 durch PKA-C](https://thumb-eu.123doks.com/thumbv2/1library_info/3651450.1503322/47.892.252.622.405.698/autoradiogramme-verdrängung-fitc-conh-kemptide-adenosin-triphosphat-phosphorylierung.webp)
![Abb. 20: Autoradiogramme der Verdrängung von [γ- 32 P]-ATP durch ATP und P 3 -(N ε -(N-TRITC-[K5, V6, A7]- A7]-CONH 2 -Kemptide)-lysyl)-P 1 -adenosin-triphosphat 3 bei der Phosphorylierung von c-C1C2 durch PKA-C](https://thumb-eu.123doks.com/thumbv2/1library_info/3651450.1503322/48.892.206.663.135.454/autoradiogramme-verdrängung-tritc-conh-kemptide-adenosin-triphosphat-phosphorylierung.webp)
![Abb. 27: Autoradiogramme der Verdrängung von [γ- 32 P]-ATP durch ATP und P 3 -(4-(N-FITC-Aminoethyl)- -(4-(N-FITC-Aminoethyl)-phenylamino)-P 1 -adenosin-triphosphat 19 bei der Phosphorylierung von c-C1C2 durch PKA-C](https://thumb-eu.123doks.com/thumbv2/1library_info/3651450.1503322/56.892.217.660.693.1042/autoradiogramme-verdrängung-aminoethyl-aminoethyl-phenylamino-adenosin-triphosphat-phosphorylierung.webp)
ÄHNLICHE DOKUMENTE
Lavogina D., Lust M., Viil I., König N., Raidaru G., Rogozina J., Enkvist E., Uri A., Bossemeyer D., Structural Analysis of ARC-Type Inhibitor (ARC-1034) Binding to Protein Kinase
Der großen Diversität der antigenspezifischen Rezeptoren und Antikörper des erworbenen Immunsystems liegen genetische Rekombinationsmechanismen
Mögliche Auswirkungen des inflammatorischen Milieus auf Proteinebene in Myotuben wur- den mit immunzytochemischen Nachweismethoden untersucht. Hierzu wurden die pri- mären
Kinetic parameters, characterizing interaction of the bifunctional inhibitor AdoC(Ahx)Arg 6 with protein kinase A in the presence of two substrates, ATP and Kemptide,
ROS are thought to promote atherosclerosis through a variety of mechanisms, including enhanced oxidation of lipoproteins (Steinberg 1997), activation of proinflammatory genes (Marui,
Als alleinige Methode für die Aufreinigung von binären oder multimeren Protein-Protein Komplexen ist die Co-Immunpräzipitation eher ungeeignet, da zum einen hochaffine
Mittels Bindung am Eisen der Hämgruppe und nachfolgender Oxidation zu Fe 3+ wird die Bindung von NO an das Eisen der Hämgruppe und die Aktiverung der löslichen
Des Weiteren konnten für diese Aminosäuren sowohl eine Auto-Phosphorylierung als auch eine Phosphorylierung durch eine andere Kinase gezeigt werden, die bei besserer Auflösung
