Ab Initio Molecular Dynamics: Proton Transport In Water
12
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
Abbildung
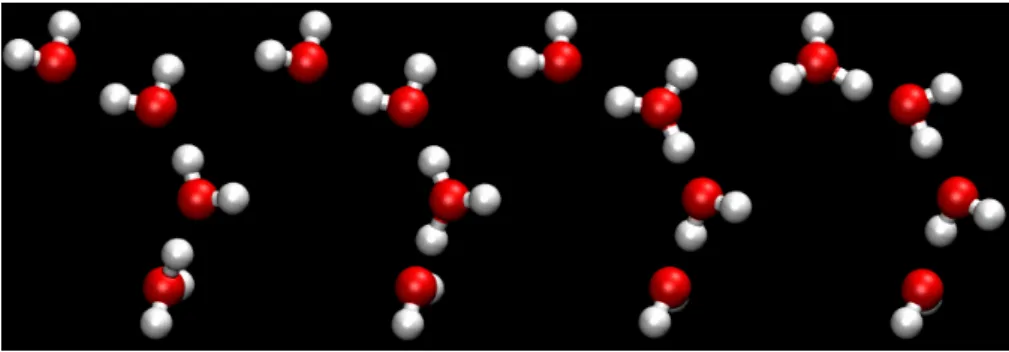


+4

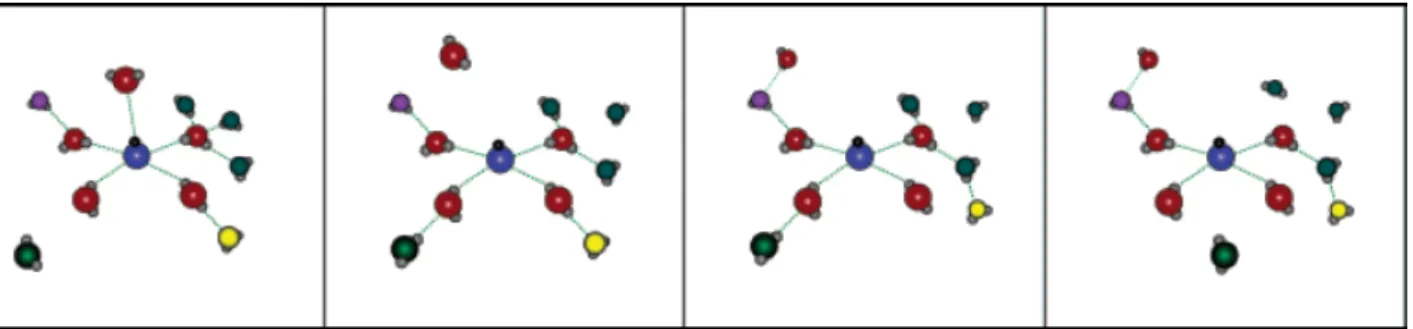
ÄHNLICHE DOKUMENTE
Having recognized this, we should, in principle, be able to "starve" an entire mode of organization (i.e., a style of problem solving) out of existence merely by reducing
A host of researchers in the last 15 years [8] have suggested another way to explain software architectures: Instead of pre- senting an architectural model as a
The work, conceived as a pilot project, which could be extended and repeated in other areas, is based on an empirical research, done by a visual check of what is identifiable
“to ensure the transition to democracy, including through promoting, facilitating and providing technical advice and assistance to a single, inclusive and transparent
The Fethullah Gülen Movement in the Con- text of Turkey’s Socio-structural Development Although the manner in which the Fethullah Gülen movement, its emergence and evolution,
Arab spring protests that began in Tunisia with a somewhat Algerian songbook quickly spread back to Algeria, which then saw more self-immolations than any other Arab
The authors detail the problems in four domains – (i) funding of teaching and research; (ii) the ‘market’ for political science education; (iii) career development, salaries, and
Tick what happens. Bridget and Annie are watching TV. The phone rings. It’s a call for someone called Rock Thrust. Two members of the FBI come into the flat. Bridget gets angry
