DISS. ETH NO.26930
Development of myo-inositol hexakisphosphate analogues as inhibitors of kidney calcification
A thesis submitted to attain the degree of DOCTOR OF SCIENCES of ETH ZURICH
(Dr. sc. ETH Zurich)
presented by
Anna Kletzmayr Master of Science Uppsala University, Sweden
born on 11.06.1993 citizen of Austria
Examiners
Prof. Dr. Jean-Christophe Leroux Prof. Dr. Robert Grass
2020
TABLE OF CONTENT
_____________________________________________________________
Table of content
Table of content ... 2
Summary ... 4
Zusammenfassung ... 6
Chapter 1: Background and purpose ... 8
1.1 Renal calcification disorders ... 9
1.2 Common pathomechanism of calcium crystal-induced kidney injury ... 10
1.3 Myo-inositol hexakisphosphate (IP6) analogues as novel treatment option for calcification disorders .... 11
1.4 Scope of this thesis ... 12
Chapter 2: Investigational therapies for primary hyperoxaluria ... 13
2.1 Introduction ... 14
2.2 Targeting the underlying molecular defect in the liver ... 17
2.3 Prevention of CaOx-induced kidney damage ... 22
2.4 Oxalate degradation in the intestine as a therapeutic modality in PH ... 26
2.5 Concluding remarks ... 27
Chapter 3: Implications of renal CaP crystallization in kidney diseases ... 29
3.1 Motivation ... 30
3.2 The role of phosphate in CKD progression ... 30
3.3 CaP as an initiator of calcium kidney stone formation ... 32
3.4 Image-based analytics for drug screening ... 33
3.5 Calcium imaging modalities ... 35
3.6 Concluding remarks ... 36
Chapter 4: Inhibitors of calcium oxalate crystallization for the treatment of oxalate nephropathies ... 37
4.1 Introduction ... 38
4.2 Materials and Methods ... 39
4.3 Results ... 46
4.4 Discussion ... 59
Chapter 5: Development of a kidney calcification inhibitor employing image-based profiling: a proof-of- concept study ... 65
5.1 Introduction ... 66
5.2 Materials and Methods ... 67
TABLE OF CONTENT
_____________________________________________________________
5.4 Discussion ... 87
Chapter 6: Conclusions and outlook ... 89
6.1 A library of IP6 analogues adaptable to different types of renal calcification ... 90
6.2 Image-based toolkits for calcification disorders ... 92
Appendix ... 96
List of abbreviations ... 125
Acknowledgments ... 128
References ... 130
SUMMARY
_____________________________________________________________
Summary
The kidneys’ role in mineral homeostasis, waste removal and urine concentration make this organ susceptible to the crystallization of the filtered substances. Of particular concern is the intratubular crystallization of calcium salts which leads to nephrocalcinosis, kidney stone formation, acute kidney injury and chronic kidney disease (CKD).
Underlying conditions triggering renal calcium crystallization in the form of calcium phosphate (CaP) or calcium oxalate (CaOx) are diverse and range from rare genetic disorders to common diseases. Prevalence of kidney stones, a common kidney calcification disorder, reaches up to 15% in high income countries. Incidence rates of kidney calcification have been sharply rising in the last decade, which can be related to changes in lifestyle and diet, hence highlighting the susceptibility of the kidney to systemic influences. Further amplifying the problem are the severe consequences of kidney calcification disorders, namely an increased risk of progressive CKD, end stage renal disease, hypertension and cardiovascular calcification. Together, kidney calcification significantly burdens the patient and the healthcare system.
Despite the high unmet medical need no renal calcification inhibiting drugs are commercially available. Their development is hampered by limited knowledge on disease pathomechanisms of a broad range of disorders and difficulties in patient diagnosis and stratification. In this thesis, we aimed at investigating myo-inositol hexakisphosphate (IP6) based analogues as renal calcification inhibitors that can be adaptable to different types of kidney calcification. To meet this objective, we established image-based tools to accelerate the pre-clinical drug discovery process in the field of calcification disorders.
In chapter 1, an overview of the broad range of kidney calcification disorders is given.
Additionally, pathophysiology and treatment options are briefly summarized.
A more detailed review of therapeutic modalities for primary hyperoxaluria (PH), a rare genetic disorder characterized by severe renal CaOx crystallization, is provided in chapter 2.
The broad range of treatment approaches currently tested in pre-clinical research and clinical trials is described.
Chapter 3 reviews the role of CaP in kidney disease. Our interest focuses on recent research highlighting its initiating role of calcium kidney stones and its driving role in CKD. Image-based drug discovery strategies that can be applied to explore kidney calcification in vitro in the absence of specific biomarkers are discussed.
In chapter 4, the development of renal CaOx crystallization inhibitors is presented. An image- based assay to assess CaOx crystalluria was implemented. Mechanistic insights gained from the screening of a small library of previously established IP6 analogues supported the design
SUMMARY
_____________________________________________________________
A selected lead compound was shown to have in vitro and in vivo efficacy to prevent CaOx crystallization and the associated kidney damage.
In chapter 5, the broader applicability of the IP6 analogue library for the inhibition of renal CaP or CaP-induced CaOx crystallization was explored. Because of the limited knowledge on the pathophysiology of kidney calcification, an in vitro calcification profiling platform was established. The platform allowed the rapid testing of varying ionic conditions and the quantification of a multitude of single cell and calcium deposit features. Leveraging the previously established library of IP6 analogues, a renal CaP inhibitor was identified, which showed in vitro and in vivo efficacy to reduce CaP-induced kidney tissue damage.
In chapter 6, the main findings of the thesis are discussed. An outlook on the expected challenges for the clinical development of the identified inhibitors is given. Further, broader applications of the established image-based toolkits for calcification disorders are suggested.
ZUSAMMENFASSUNG
_____________________________________________________________
Zusammenfassung
Die Rolle der Nieren bei der Mineralhomöostase, der Abfallentfernung und der Urinkonzentration macht das Organ anfällig für die Kristallisation der gefilterten Substanzen.
Aus medizinischer Sicht besonders problematisch ist die intratubuläre Kristallisation von Kalziumsalzen, die zu Nephrokalzinose, Nierensteinbildung, akutem Nierenschaden und chronischer Nierenerkrankung führt.
Die zugrunde liegenden Bedingungen, die eine renale Calciumkristallisation in Form von Calciumphosphat (CaP) oder Calciumoxalat (CaOx) auslösen, sind vielfältig und reichen von seltenen genetischen bis hin zu häufigen vorkommenden Krankheiten, zum Beispiel Nierensteinen. Die Prävalenz von Nierensteinen erreicht in Industriestaaten bis zu 15%. Die Inzidenzraten der Nierenverkalkung sind im letzten Jahrzehnt stark gestiegen, dies kann, unter anderem, mit Änderungen des Lebensstils und der Ernährung zusammenhängen. Damit wird die Anfälligkeit der Niere für systemische Einflüsse nochmals deutlich hervorgehoben. Eine weitere Verstärkung des Problems sind die schwerwiegenden Folgen von Nierenverkalkungsstörungen, nämlich ein erhöhtes Risiko für fortschreitendes, chronisches Nierenversagen, Bluthochdruck und kardiovaskuläre Verkalkung. Die hohe Prävalenz zusammen mit den drastischen gesundheitlichen Auswirkungen von Nierenverkalkungen belastet Patienten und das Gesundheitssystem erheblich.
Trotz des hohen medizinischen Bedarfs sind keine Medikamente zur Hemmung der Nierenverkalkung in klinischem Gebrauch. Begrenzte Kenntnisse über die Pathophysiologie der weiten Bandbreite von Nierenerkrankungen und Probleme bei der Diagnose und Stratifizierung von Patienten erschweren die Entwicklung von Inhibitoren. Das Ziel dieser Arbeit war die Entwicklung von myo-inositolhexakisphosphat (IP6) Analoga als Inhibitoren der Nierenverkalkung. Die Moleküle sollen, je nach Art der Nierenverkalkung, adapiert werden können und damit die breite Bandweite von Nierenverkalkungskrankheiten abdecken. Um dieses Ziel zu erreichen, haben wir zudem bildbasierte Methoden entwickelt, um den vorklinischen Entwicklungsprozess im Bereich der von Verkalkungskrankheiten zu beschleunigen.
In Kapitel 1 wird ein Überblick über das breite Spektrum von Nierenverkalkungsstörungen gegeben. Zusätzlich werden Pathophysiologie und Behandlungsmöglichkeiten kurz zusammengefasst.
Eine detailliertere Übersicht über Therapiemöglichkeiten der Primären Hyperoxalurie (PH), eine seltene genetische Störung, die durch eine schwere renale CaOx-Kristallisation gekennzeichnet ist, wird in Kapitel 2 gegeben. Das breite Spektrum der derzeit in präklinischen und klinischen Studien getesteten Behandlungsansätze wird beschrieben.
ZUSAMMENFASSUNG
_____________________________________________________________
Nierensteinen und dessen Rolle in fortschreitendem chronischem Nierenversagen hervorheben. Es werden bildbasierte Strategien zur Wirkstoffentdeckung diskutiert, mit denen eine Vielzahl von Nierenverkalkungsprozessen in vitro ohne genaue Vorkenntnisse über spezifische pathophysiologische Vorgänge untersucht werden könnten.
In Kapitel 4 wird die Entwicklung von Inhibitoren der renalen CaOx-Kristallisation präsentiert.
Eine bildbasierter Methode zur Quantifizierung der CaOx-Kristallurie wurde implementiert.
Mechanistische Erkenntnisse aus dem Testen einer Reihe zuvor etablierter IP6-Analoga unterstützten das Design neuartiger, hochwirksamer di- und dreiwertiger myo- inositolpentakisphosphat (IP5) Moleküle. Ein ausgewähltes IP5 Molekül wurde in vitro und in vivo getestet und zeigte Wirksamkeit in der Inhibierung von CaOx-Kristallisierung und der damit verbundene Nierenschädigung.
In Kapitel 5 wurde die breitere Anwendbarkeit der IP6-Analogserie zur Hemmung der renalen CaP- oder CaP-induzierten CaOx Kristallisierung untersucht. Aufgrund des begrenzten Wissens über die Pathophysiologie der Nierenverkalkung wurde eine Plattform zur Profilierung von in vitro Kalzifizierungsprozessen entwickelt. Die Plattform ermöglichte das schnelle Testen unterschiedlicher Kalzifizierungsbedingungen und die Quantifizierung einer Vielzahl von Zell- und Kalziumsablagerungsmerkmalen. Aus der zuvor etablierten Reihe von IP6-Analoga wurde ein renaler CaP-Inhibitor identifiziert, der in vitro und in vivo Wirksamkeit zur Verringerung von CaP-induzierten Nierenschäden zeigte.
In Kapitel 6 werden die wichtigsten Ergebnisse der Arbeit diskutiert. Es wird ein Ausblick auf die zu erwartenden Herausforderungen für die klinische Entwicklung der identifizierten Inhibitoren gegeben. Darüber hinaus wird die breitere Anwendung der etablierten bildbasierten Methoden im Bereich von Organkalzifizierungen erwogen.
CHAPTER 1: BACKGROUND AND PURPOSE
_____________________________________________________________
Chapter 1: Background and purpose
CHAPTER 1: BACKGROUND AND PURPOSE
_____________________________________________________________
1.1 Renal calcification disorders
Due to the role of the kidney in mineral secretion and urine concentration, this organ is highly susceptible to crystallization of various substances1. Calcium crystallization in the form of calcium phosphate (CaP) and calcium oxalate (CaOx) is a major type of renal crystallization, causing kidney stone formation (urolithiasis), diffuse calcification of the renal parenchyma (nephrocalcinosis), acute kidney injury (AKI) and chronic kidney disease (CKD)1,2. While it is difficult to estimate the total incidence of kidney calcification disorders, prevalence for kidney stones and CKD reaches up to 15% each in high-income countries2–4. Numbers have been sharply rising within the last twenty years5, which could, at least in part, be attributed to changes in lifestyle and dietary habits2. Risk factors for renal calcification include obesity, hypertension and diabetes6,7, further emphasizing the influence of lifestyle and diet. Vice versa, kidney stone patients are at increased risk of developing hypertension8. Kidney calcification invariably damages the organ and thus favours progressive CKD and end-stage renal disease (ESRD)9–11. The high prevalence and serious outcomes of kidney calcification disorders highlight the need for better preventive, diagnostic and therapeutic options.
A broad range of underlying disorders or conditions causes renal calcium crystallization (Table 1.1). For these conditions, a classification has been proposed, consisting of two partially overlapping groups – acute and chronic exposure to crystals causing AKI or CKD, respectively1. In the first case, inflammatory bowel disease, intestinal bypass surgery, and drugs altering intestinal absorption are underlying conditions that can trigger an increased oxalate uptake from the ileum or colon, which in turn can cause hyperoxaluria and CaOx crystallization12–14. Moreover, drug exposure, toxins or dietary components can directly elicit hyperoxaluria, for example via high intake of oxalate-rich foods, vitamin C supplements or poly(ethylene glycol) intoxication15–17. High oral sodium phosphate intake, through e.g. oral bowel purgatives, can drive renal CaP precipitation and acute phosphate nephropathy18. In the second case, chronic exposure is mostly caused by genetic disorders, disturbing mineral metabolism or renal mineral transport. Diseases include primary hyperoxaluria (PH), genetic forms of renal tubular acidosis or Dent’s disease, which trigger CaOx and/or CaP crystallization within the tubules10,19,20. For both acute and chronic conditions, the development of kidney stones is reported10,12,17. Kidney stones cause organ injury by physical obstruction of tubules, ureter or bladder2. Of note, the largest population of kidney stone patients does not show any underlying disorders2,12. Additionally, CaP-induced kidney injury could ensue as a result of nephron loss and hence increased phosphate per nephron secretion, occurring in ageing or in early-stage CKD21,22. Thereby, CaP is suggested to be an important driver of CKD progression23. The role of CaP in CKD progression and idiopathic kidney stone formation is discussed in detail in chapter 3.
CHAPTER 1: BACKGROUND AND PURPOSE
_____________________________________________________________
Table 1.1: A non-exhaustive overview of calcium crystal nephropathies1,12,14,23,24. Disease/Stimuli Subtypes Crystal
type Renal
damage Treatment/Management Genetic
diseases Primary hyperoxaluria CaOx CKD Increase fluid intake, specific interventions to reduce mineral or metabolite excretion Genetic forms of renal
tubular acidosis CaOx, CaP CKD Dent disease, Lowe
syndrome, idiopathic infantile
hypercalcaemia and hypercalciuria
CaP CKD
Enteric crystal
nephropathy Bariatric surgery, inflammatory bowel disease, short bowel syndrome
CaOx AKI Avoid dietary oxalate
intake, increase fluid intake, treat enteric disease Diet-induced
crystal nephropathy
Diet rich in oxalate (spinach, rhubarb) or phosphate (processed foods with phosphate containing
preservatives)
CaP, CaOx AKI Stop exposure, increase fluid intake
Drug-induced crystal nephropathy
Orlistat, oral bowel
purgatives CaP, CaOx AKI Stop exposure, increase fluid intake
Idiopathic
calcium stones CaOx, CaP CKD Surgical removal, increase
fluid intake, dietary recommendations,
thiazide-type diuretics, potassium alkali
Nephron loss Ageing, early-stage CKD CaP CKD Increased fluid intake, dietary recommendations, phosphate binders
1.2 Common pathomechanism of calcium crystal-induced kidney injury
Urinary supersaturation with respect to calcium, phosphate and/or oxalate leads to precipitation of CaP or CaOx within the renal tubules25–27. Subsequently, intratubular crystals adhere to the renal epithelium through crystal adhesion proteins, such as annexins or osteopontin, causing nephrocalcinosis28,29. Rapid and diffuse crystallization triggers direct crystal-induced cytotoxicity, suggested to involve necroptosis, a form of regulated necrosis30. Necrotic cells directly elicit inflammation through the release of damage-associated molecular patterns31. Upon chronic exposure, intratubular crystals might also be translocated to the interstitial tissue, as a mechanism of crystal clearance32,33. In the interstitial tissue, macrophages are involved in the removal of crystals34. Regardless of the underlying cause,
CHAPTER 1: BACKGROUND AND PURPOSE
_____________________________________________________________
proposed to play a central role in mediating the progressive renal failure in various crystal- induced nephropathies1,35–38.
1.3 Myo-inositol hexakisphosphate (IP6) analogues as novel treatment option for calcification disorders
Kidney calcification is driven by urinary supersaturation with respect to calcium, phosphate and/or oxalate. Hence, an overarching treatment recommendation is the increased fluid intake to reduce urinary supersaturation2,10,12. In a large subset of calcium crystal nephropathies, such as enteric and diet-induced crystal nephropathies, dietary recommendations might be beneficial to reduce crystallization13,17,39. A reduced protein, low salt, normal calcium diet was suggested to lower the risk of calcium stone recurrence compared to a low calcium diet39. Treatment guidelines for CKD patients include recommendations to restrict phosphate intake, and the administration of phosphate binders to reduce serum phosphate concentrations40,41. Existing kidney stones can be removed by minimally invasive endourological treatments or shockwave lithotripsy. Preventive measures include thiazide diuretics in case of hypercalciuria and urine alkalizing agents against CaOx crystallization2. However, caution has to be taken with alkalizing agents, as with increasing urinary pH, CaP precipitation is favored2,12. Correction of causal genetic or metabolic abnormalities can present a treatment possibility with certain underlying diseases10, which is discussed in detail in chapter 2 for PH.
Overall, current treatment options are insufficient to prevent CKD progression and ESRD development in severe cases of kidney calcification10. Hence, a renal calcium crystallization inhibitor would be of tremendous value as a common treatment option for renal calcification disorders that acts independently of the underlying cause.
Previously, IP6, ubiquitously found in eukaryotic cells42, has been shown to be an effective inhibitor of calcium crystallization43,44. So far, its application as a calcium crystallization inhibitor in various crystallization disorders has been hampered by its rapid dephosphorylation in plasma. Urinary exposure of orally administered IP6 was found to be minimal, thus making it unsuitable as a renal calcium crystallization inhibitor45. Stability in plasma and calcium-chelating properties of the molecule were tuned by substituting phosphate groups with different anionic groups or oligoethylene glycol (OEG)46. Further, enhanced inhibition of CaP crystallization in serum and vascular system was achieved with such an IP6 analogue47. Thus, we hypothesize, that leveraging the established small library of IP6 analogues could provide a promising starting point for the development of renal calcification inhibitors.
CHAPTER 1: BACKGROUND AND PURPOSE
_____________________________________________________________
1.4 Scope of this thesis
Due to the well-established pathological role of CaOx crystallization in severe genetic disorders and their predominant presence in calcium kidney stones, the primary aim of this work was the development of IP6 analogues as inhibitors of renal CaOx crystallization. In a second step, their potential application as a renal CaP or dual CaP-CaOx inhibitor was further explored, which would allow their broad applicability in a wide range of kidney calcification disorders. Inhibitor development is hampered by limited knowledge on the pathomechanims of a diverse set of diseases and problems concerning patient diagnosis, stratification and treatment response. Hence, in the course of the work we developed image-based methods to accelerate discovery in the field of kidney calcification.
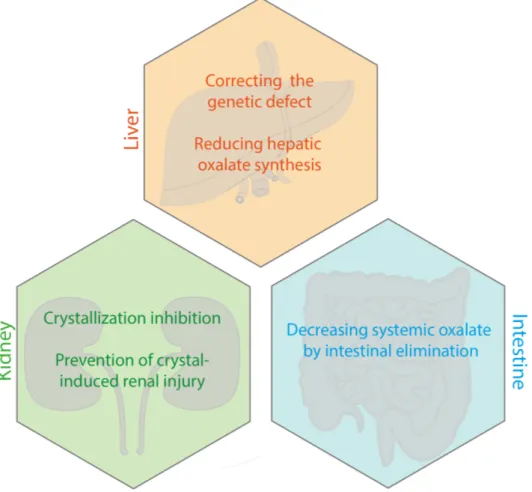
In chapter 2, novel treatment modalities for PH were reviewed. We explain current treatment recommendations and therapeutic approaches currently in clinical trials or pre-clinical development. Strategies include the correction of the underlying genetic defect or metabolic abnormalities in the liver, kidney-protective measurements and reducing the systemic oxalate load via increasing intestinal elimination.
The focus of chapter 3 lies on extending the application of IP6 analogues towards renal CaP and dual CaP-CaOx crystallization inhibition. Implications of CaP in CKD progression, as well as in the formation of kidney stones are reviewed. Due to the limited current knowledge on the multifactorial process of kidney calcification, involving both crystal formation and crystal- induced tissue injury, we discuss potential strategies of image-based calcification profiling to accelerate discoveries in this field.
In chapter 4, the assessment of novel IP6 analogues as renal CaOx inhibitors is presented.
Herein, we show the design of novel, highly efficient IP6 analogues that inhibit CaOx crystallization, as well as crystal-induced cell injury in vitro and in vivo.
In chapter 5, we established an image-based calcification profiling tool to accelerate pre- clinical drug discovery in the field of calcification disorders. We then leveraged the previously established library of IP6 analogues to identify a renal CaP or dual CaP-CaOx inhibitor. The lead compound demonstrated in vitro and in vivo efficacy to prevent CaP-induced kidney damage.
In chapter 6, we discuss the main findings and provide an outlook on the challenges lying ahead in the clinical development of the herein identified kidney calcification inhibitors. We further give an outlook on the broader applicability of the image-based toolkits established as part of this work.
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
Chapter 2: Investigational therapies for primary hyperoxaluria
This chapter was adapted from the published paper: A. Kletzmayr, M. E. Ivarsson and J.-C. Leroux, Investigational therapies for primary hyperoxaluria, Bioconjugate Chem., 2020, https://doi.org/10.1021/acs.bioconjchem.0c00268.
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
2.1 Introduction
PH is a group of autosomal recessive diseases, which is characterized by high endogenous production of oxalate. PH is a rare disorder, with an estimated prevalence of 1-3 cases per million population, and is associated with devastating consequences for the patient, posing a great challenge for disease management10,48,49. Enzymatic deficiencies in the hepatic glyoxylate metabolism are the underlying cause leading to increased oxalate production. The excess oxalate is eliminated through the kidneys to keep plasma oxalate levels within normal limits (< 10 µmol/L)10. Hyperoxaluria occurs when the urinary secretion of oxalate shows continuously elevated levels of > 0.7 mmol/1.73 m2/day10. In the renal tubules, increasing concentrations of oxalate trigger the crystallization of calcium oxalate, leading to urolithiasis and nephrocalcinosis10,50. Additionally, CaOx crystals elicit an inflammatory response and fibrosis of the tissue contributing to progressive renal damage1,30,37. The sustained kidney damage can progress to CKD and ESRD, ultimately leading to a need for renal replacement therapy10,50. Declining renal function causes reduced excretion of oxalate and further buildup of oxalate in the kidneys and other tissues, creating a vicious circle that inexorably drives loss of kidney function10,50.
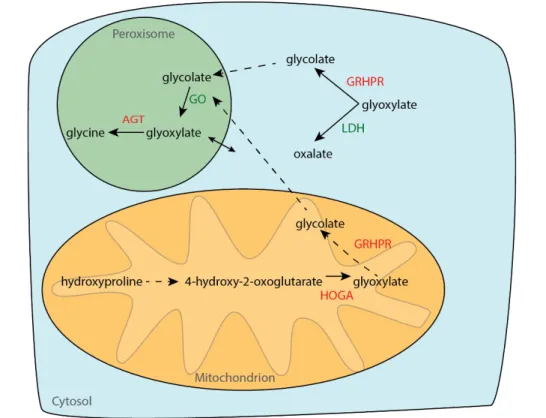
PH is divided into three subtypes, depending on the gene affected by mutations. The most common and severe form is PH type 1, accounting for 70 – 80% of PH cases worldwide. In PH1, mutations in the gene encoding for the liver specific peroxisomal enzyme alanine-glyoxylate aminotransferase (AGT) lead to the accumulation of the oxalate precursor glyoxylate (Fig. 2.1).
Cytosolic glyoxylate is metabolized by lactate dehydrogenase (LDH) causing the overproduction of oxalate10. More 150 different mutations for the AGT gene have been identified, affecting AGT function by, for example, accelerating enzyme degradation, impairing correct enzyme folding, or causing mistargeting from its normal location in the peroxisome to mitochondria51,52. PH type I poses a high risk for ESRD, with up to 20 % of children even reaching this stage within the first year of life50. PH types 2 and 3 are less common, and are characterized by mutations in the genes encoding for glyoxylate reductase/hydroxypyruvate reductase and 4-hydroxyl-2-oxoglutarate aldolase, respectively (Fig. 2.1)10,50. glyoxylate reductase/hydroxypyruvate reductase mutations lead to accumulation of its substrate glyoxylate, which is converted to oxalate by LDH. Mechanisms by which HOGA mutations cause hyperoxaluria are still unkown. PH2 was generally assumed to show a more benign disease course than PH1, being associated with recurrent urolithiasis, but less profound hyperoxaluria and nephrocalcinosis than PH1. However, a recent retrospect study highlights significant renal impairment in PH2 patients, low transplant success rates and high mortality rates53. PH3 is considered the least critical subtype, with no reports of ESRD50.
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
Fig. 2.1: Schematic of the hepatic glyoxylate metabolism. (AGT: alanine-glyoxylate aminotransferase, GRHPR: glyoxylate reductase/hydroxypyruvate reductase, HOGA: 4-hydroxyl-2-oxoglutarate aldolase, GO: glycolate oxidase, LDH: lactate dehydrogenase)10,50..
Few supportive measures are available to slow down disease progression towards ESRD.
Those include high fluid intake, restricted oxalate consumption and administration of oral citrate as a urine alkalizing agent aiming to reduce CaOx crystallization10,50. Kidney stones can be surgically removed2. Pyridoxine supplementation has shown positive effects in a subset of PH1 patients with residual AGT activity54,55. Pyridoxine is the precursor of pyridoxal phosphate, which acts as a co-factor for the defective enzyme, increasing AGT activity and peroxisomal targeting56. Neither hemodialysis nor peritoneal dialysis, are efficient enough to reduce plasma oxalate levels once they have become elevated. Thus, hemodialysis should be initiated early and the period until transplantation should be kept short to prevent systemic overload. Frequent, but shorter hemodialysis sessions (e.g. , 5 – 6 times per week over 3 hours) are recommended50. To date, organ transplantation is the only available curative treatment for PH patients. In most cases, combined liver-kidney transplantation is the methodof choice, to correct both the genetic defect in the liver and the CaOx-induced kidney damage50. For a more detailed discussion of epidemiology, clinical spectra, diagnosis and treatment of PH the interested reader is referred to other in-depth reviews on those topics10,50.
Advances in gene therapy, siRNA approaches and probiotics have fostered the investigation of different strategies to reduce the excess systemic oxalate levels in PH57–60. Additionally, an increasing understanding of crystal handling by the kidney has highlighted potential drug targets in the kidney1,61,62. While such efforts have already resulted in a few compounds being
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
studied in clinical trials up to phase 3 (Table 2.1), insights from basic research and pre-clinical studies reveal exciting new and revisited targets. This review is focused on advances in both basic and clinical research, into investigational treatment approaches for PH, ranging from targeting molecular defects in the liver, to fighting kidney damage and stimulating intestinal elimination of oxalate (Fig. 2.2).
Fig. 2.2: Overview of investigated treatment strategies in PH.
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
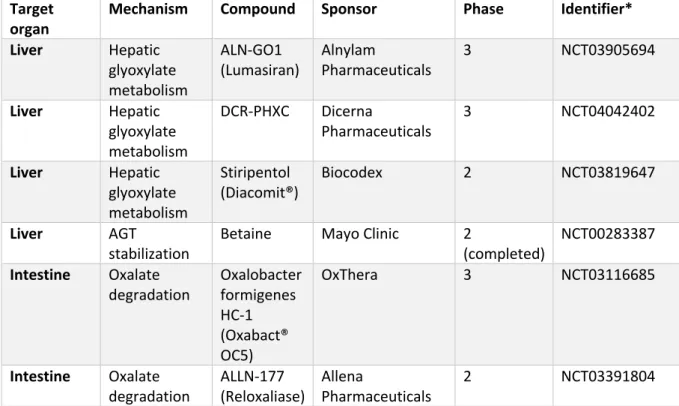
Table 2.1: Investigational new treatments for primary hyperoxaluria currently in clinical trials.
Target
organ Mechanism Compound Sponsor Phase Identifier*
Liver Hepatic glyoxylate metabolism
ALN-GO1
(Lumasiran) Alnylam
Pharmaceuticals 3 NCT03905694
Liver Hepatic glyoxylate metabolism
DCR-PHXC Dicerna
Pharmaceuticals
3 NCT04042402
Liver Hepatic glyoxylate metabolism
Stiripentol
(Diacomit®) Biocodex 2 NCT03819647
Liver AGT
stabilization Betaine Mayo Clinic 2
(completed) NCT00283387 Intestine Oxalate
degradation Oxalobacter formigenes HC-1 (Oxabact®
OC5)
OxThera 3 NCT03116685
Intestine Oxalate degradation
ALLN-177 (Reloxaliase)
Allena
Pharmaceuticals
2 NCT03391804
*Details about the clinical trials can be found using the NCT number on https://clinicaltrials.gov/. The data were retrieved on the 29th of April 2020.
2.2 Targeting the underlying molecular defect in the liver
2.2.1 Gene and cell therapies
PH is a monogenic liver disease, thus one of the most direct therapeutic approaches comprises strategies to correct the hepatic alterations in glyoxylate metabolism. Both gene and cell therapy approaches have been assessed to either correct the genetic defect or replace mutated hepatocytes with healthy cells50,59,63. However, in contrast to many other genetic diseases, PH symptoms are caused by the accumulation of a metabolic product (oxalate) rather than a deficiency or absence of a certain protein10. Hence, correcting the function of a majority of hepatocytes is required to achieve a therapeutic effect. Standard hepatocyte transplantation procedures are insufficient to ensure a high enough fraction of delivered cells51,64. A few pre-clinical studies have evaluated the effects of preparative regimes to provide a proliferation advantage of transplanted over host cells. Hepatic irradiation was employed to reduce host cell proliferation in combination with viral delivery of growth factors stimulating transplanted cells63,65. Two studies have reported engraftment of transplanted cells with this approach and concurrent reduction in urinary oxalate excretion in an Agxt-/- mouse model63,65. Importantly, the treatment prevented nephrocalcinosis induced by ethylene glycol challenge, a precursor of glycolate triggering increased oxalate production and renal CaOx deposition, suggesting sufficient cell engraftment63. Nevertheless, safety concerns regarding the viral delivery of hepatocyte growth factor and side effects of the irradiation
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
procedure persist65. Additionally, the source of healthy hepatocytes in practice would need to be clearly defined and potential immunogenic reactions considered66,67. One clinical case report of liver cell transplantation in a PH1 patient with severe systemic oxalosis indicated positive effects of cell transplantation, suggesting it as a potential bridging procedure until organ transplantation. In this case, a total of 0.3 x 109 hepatocytes/kg were administered in daily infusions over six consecutive days via a catheter placed in the portal vein. The patient condition, with regard to plasma oxalate levels and clinical status (e.g., weight gain), markedly improved over eight months allowing for combined liver–kidney transplantation. Still, long- term engraftment of a high percentage of healthy donor cells, as well as the need for immunosuppressive therapy present major hurdles for cell therapy in PH68.
Correction of the mutant gene has also been reported, using both in vivo and ex vivo gene correction strategies. On the one hand, in vivo correction of the AGT gene was studied in the Agxt-/- mouse model employing adeno-associated or helper-dependent adenoviral vectors.
Sustained transgene expression and correction of the PH1 phenotype, meaning a decrease in urinary oxalate levels, over 7 to 28 weeks was achieved. However, while both types of vectors could provide long term expression69, limitations regarding acute toxicity and immunogenicity due to the high virus doses needed to achieve sufficient correction still have to be faced59,70. On the other hand, ex vivo gene modification using patient-derived cells was also studied.
Induced pluripotent stem cells were generated from PH1 donors, which were subsequently corrected by lentiviral delivery of wild-type AGT and differentiated towards hepatocyte-like cells in vitro. While rescue of AGT protein expression in the generated hepatocyte-like cells was confirmed, no data on their behavior in an animal disease model has been reported. Thus, this strategy as therapeutic avenue still needs to be validated66. Nevertheless, the approach is noteworthy because it opens opportunities for in vitro disease modeling and gene therapy experiments by enabling the generation of hepatocyte-like cells with patient-specific genetic backgrounds.
2.2.3 Reducing oxalate synthesis by inhibition of hepatic glyoxylate metabolism While directly correcting the disease-causing gene in PH has proven to be difficult in in vivo studies due to the need of altering a high number of cells, reducing the hepatic oxalate synthesis by the inhibition of enzymes involved in the production of oxalate presents a more feasible therapeutic approach (Fig. 2.1). As described below, siRNA, CRISPR-Cas9 or small molecule strategies are being investigated to reduce expression or inhibit catalytic activity of enzymes involved, ultimately resulting in a decrease in oxalate production58,71,72. The value of this targeting approach is supported by a recent clinical study, that revealed a correlation of urinary oxalate levels and progression towards ESRD in PH patients with advanced CKD73. Thus, decreasing urinary oxalate excretion could slow down or completely halt progression towards ESRD, even if some degree of hyperoxaluria persists.
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
2.2.3.1 Targeting glycolate oxidase expression in PH1
In the hepatic metabolism of glyoxylate, glycolate oxidase (GO) which is encoded by the hydroxyacid oxidase 1 (HAO1) gene, catalyzes the conversion of glycolate to glyoxylate, which, in healthy individuals, is further metabolized to glycine by AGT (Fig. 2.1)50. In the case of PH1, AGT mutation leads to glyoxylate accumulation, which is further converted to oxalate by lactate dehydrogenase (LDH), causing the excess production of oxalate50. Hence, GO was proposed as a target in PH based on the hypothesis that less glyoxylate accumulates with GO silencing, that in turn would reduce oxalate production74. This approach would concomitantly lead to the accumulation of glycolate. The latter is highly soluble and readily excreted by the kidney74. A case report of one patient with a defective splice variant of GO showed 20-fold increased levels of urinary glycolate, which did not result in any reported symptoms75. Accordingly, GO-deficient mice displayed glycolic aciduria, but were otherwise asymptomatic74. Together those studies suggest target safety.
Two studies have reported the successful silencing of hepatic HAO1 using siRNAs. Hepatic targeting was achieved by either a lipid nanoparticle formulation or N-acetylgalactosamine functionalization (for uptake by hepatocytes via the asialoglycoprotein receptor)58,76. Both approaches resulted in rapid and durable suppression of liver HAO1 mRNA expression in healthy mice and monkeys58,76. More specifically, in healthy mice the nanoparticle formulation showed 90% reduction in hepatic HAO1 mRNA within 12 h of 0.3 or 1 mg/kg siRNA intravenously (i.v.) administration and sustained mRNA suppression for up to 45 days. In the hyperoxaluric Agxt-/- mouse model, a single dose of 0.3 mg/kg siRNA i.v. could reduce urinary oxalate levels to < 50% up to day 15, returning back to baseline at day 28 post injection. When Agxt-/- mice were further challenged with ethylene glycol, siRNA administration at the onset of ethylene glycol challenge, day 7 and day 14 inhibited nephrocalcinosis76. Thereby, the therapeutic benefit of GO silencing was demonstrated. Importantly, no adverse effects of the treatment were detected over 150 days upon bimonthly administration of 0.3 mg/kg siRNA in rats, supporting the safety of HAO1 silencing76.
Similar effects were observed with HAO1-targeting siRNA that was functionalized with N- acetylgalactosamine (ALN-GO1). In this study, 3 mg/kg ALN-GO1 subcutaneously (s.c.) administered could suppress target mRNA expression to < 50% for up to 42 days in healthy mice. In a rat disease model, 3 mg/kg siRNA s.c. could further reduce ethylene glycol-induced hyperoxaluria by 98%58. Phase 1/2 clinical trials were successfully completed with ALN-GO1 (Lumasiran), with a reported 76% mean maximal reduction in urinary oxalate relative to baseline levels77. In December 2019, Alnylam announced positive results of a phase 3 study of Lumasiran in PH1 patients. According to the press release, both the primary efficacy endpoint, i.e., change in urinary oxalate levels from baseline, and all secondary endpoints were met78. Lumasiran received Pediatric Rare Disease Designation, Orphan Drug Designation, and Breakthrough Therapy Designation for the treatment of PH1 and a rolling submission of a new drug application has been completed by Alnylam79.
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
Recently, Zabaleta and co-workers reported GO1 disruption by employing a CRISPR-Cas9 system. They designed a single AAV8 viral vector to deliver both the Staphylococcus aureus Cas9 under a liver specific promoter and the HAO1 specific sgRNA, to Agxt-/-mice. Similarly to the siRNA approach, GO knock-out by CRISPR-Cas9 led to a decrease in urinary oxalate levels and, upon ethylene glycol challenge, reduced renal CaOx deposition 4 weeks after a single injection71. However clear benefits over the use of siRNAs for GO1 suppression were not observed. While the CRISPR-Cas9 system might alleviate the need for repeated injections by siRNAs, the potential safety issues of the viral vector used, as well as increasing chances of potential off-target effects over prolonged periods of time due to lifelong expression of the Cas9 nuclease, might, at present, overrun the patient discomfort of repeated injections.
2.2.3.2 Lactate dehydrogenase as a novel target
Recently, LDH was suggested as a more potent target than GO to reduce urinary oxalate levels57. By controlling the last step in the glyoxylate metabolism in the liver, LDH is considered a key enzyme, converting glyoxylate to oxalate (Fig. 2.1)80. LDH also controls the conversion between pyruvate and lactate in the liver and other tissues81. Deficiencies in LDH subunits were reported to affect muscle tissue function. In contrast, no liver-specific phenotypes were documented, making it an attractive target choice in PH82,83. In comparison to GO silencing, LDH targeting as a therapeutic strategy is not limited to PH1 but could be applicable to the different PH subtypes due to its control over the last, common step of hepatic oxalate production.
Comparing siRNAs targeting LDH vs. GO conjugated to N-acetylgalactosamine, Lai et al.57 reported superiority of LDH targeting in respect to reducing urinary oxalate levels in mouse models of PH, possibly because of the direct control of LDH over oxalate production. They conjugated siRNA against Ldha, the LDH gene encoding for the M subunit of the protein, with N-acetylgalactosamine and demonstrated liver-specific knock down of LDH protein expression in Agxt-/- and Grhpr-/- mouse models. Additionally, suppression of LDH mRNA expression was confirmed in chimeric mice with humanized livers and in monkeys. Importantly, no acute liver toxicity and no effects on lactate production in other tissues were observed57. The siRNA drug candidate, termed DCR-PHXC is currently in phase 3 clinical development by Dicerna (NCT04042402). According to a press release, preliminary analysis is showing normalization or near normalization of urinary oxalate levels and a favorable tolerability profile84.
Of note, a close investigation of changes in plasma and liver metabolites upon liver-specific LDH silencing in the Agxt-/- and Grhpr-/- mouse models revealed significant alterations in tricarboxylic acid cycle intermediates, lower ATP levels and lactate/pyruvate ratios85. Thus, long-term effects and species-specific differences in the hepatic oxalate metabolism still need to be evaluated and will validate the proposed superiority of LDH vs. GO targeting.
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
the potential application of stiripentol, a LDH5 inhibitor used in the treatment of Dravet syndrome, a type of epilepsy87, to reduce hepatic oxalate production. Efficacy of stiripentol was tested in two rat models of CaOx nephropathy, inducing renal CaOx crystallization by spiking of the drinking water with either ethylene glycol or hydroxyproline. The latter, similarly, to ethylene glycol, is metabolized in the liver to glyoxylate and subsequently oxalate, leading to increased urinary oxalate levels. Stiripentol was administered daily at 300 mg/kg by oral gavage for two days in the ethylene glycol model and 16 days in the hydroxyproline model. In both models, renal CaOx deposition and plasma serum creatinine, an indicator of kidney function, were significantly reduced with stiripentol treatment. Only in the hydroxyproline model, urinary oxalate levels, the most direct indication of hepatic LDH inhibition, were measured, showing a < 50% reduction between days 8 and 1572. Together, the results support activity of stiripentol on inhibiting hepatic LDH, which in turn leads to reduction in urinary oxalate, CaOx crystallization and kidney failure. Nevertheless, high doses of stiripentol administered only showed limited efficacy in the reduction of CaOx deposition and kidney injury in the hydroxyproline model. Additionally, urinary oxalate levels of Dravet patients on stiripentol treatment were analyzed and indeed showed a slight, but significant reduction in urinary oxalate levels compared to cystinuria patients that routinely have quantification of urine oxalate. While being indicative of hepatic LDH inhibitory activity in humans, it is unclear how comparable the cystinuria group was to healthy control subjects, with regard to baseline urinary oxalate levels. In a case study, stiripentol treatment at 50 mg/kg/day in a PH patient, representing the upper recommended dose limit in Dravet syndrome86, could also reduce urinary oxalate to below 50% compared to before treatment, without signs of side effects72, further suggesting potential activity of this drug in humans.
More clarity about the efficacy of stiripentol in the treatment of PH will be gained by the results of a phase II clinical study (NCT03819647). While small molecule LDH inhibitors might provide an alternative to siRNA based silencing, stiripentol is associated with important side effects, such as loss of appetite, sleep disturbance, ataxia, hyperactivity/irritability, and neutropenia, which might limit its application86,88. Additionally, it undergoes hepatic metabolism, and interacts with CYP1A2, CYP2B6 and CYP3A4, thus drug-drug interactions need to be considered86. Stiripentol has relatively low affinity for LDH586, but the forthcoming clinical trial could raise further interest in developing more potent and safer LDH5 inhibitors.
2.2.4 Mutant AGT stabilization by chemical chaperones
An estimated 30 to 50% of PH1 patients show responsiveness (as defined by a >30 % reduction in urinary oxalate) to supraphysiological doses of pyridoxine, a form of vitamin B610,48,55. Pyridoxine can act by stabilizing certain missense mutations and prevent the resulting mistrafficking of the mutant protein54,55. Ongoing investigations are focused on better understanding how the patient’s genotype affects pyridoxine response to in turn facilitate the discovery of new chemical chaperones55,89. However, in a first prospective clinical trial no conclusive results on the relationship between genotype and vitamin B6 response were obtained, potentially due to the limited number of patients enrolled55. Further, other
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
molecules, such as pyridoxal 5’-phosphate, pyridoxamine, aminooxyacetic acid and betaine, were found in in vitro studies to have stabilizing effects on different AGT mutations, and could potentially be beneficial for certain subgroups of PH1 patients89–91. Additionally, dequalinium chloride was identified to restore AGT trafficking from mitochondria to peroxisomes, leading to reduction in oxalate levels92. In a phase 2 clinical trial, oral administration of 6 – 10 mg betaine per day was tested on PH1 patients partially or non-responsive to vitamin B6 treatment. Results suggested a reduction of urinary oxalate levels upon 2 months of treatment, however only a small number of patients was enrolled and detailed results have not been published (NCT00283387). No further clinical studies with betaine have been reported since 2013. Taken together, the search for new chemical chaperones of AGT has not led to promising results so far. The poor understanding of the impact of different genetic modifications on the mutant protein together with the vast array of mutations possible, make the identification of chemical chaperones and the required patient stratification a difficult endeavor.
2.3 Prevention of CaOx-induced kidney damage
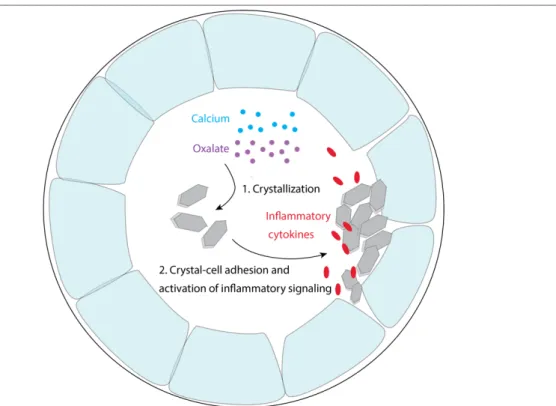
PH results from an overproduction of oxalate in the liver. Nevertheless, due to renal excretion of the excess oxalate and the resulting intratubular crystallization of CaOx, the main affected organ is the kidney10. It has been found that independent of the underlying source of crystallization, crystal nephropathies share common pathways of crystal–cell interactions, leading to kidney inflammation and injury1,30,37. Hence, the advantage of kidney targeting lies in the potential broader application range of the developed drugs, from specifically targeting PH to different types of crystal nephropathies. An increasing number of studies have recently investigated the interplay of CaOx or other types of crystals with the renal epithelium. It was suggested that, first, high urinary calcium and/or oxalate levels drive the intratubular crystallization of CaOx. Subsequently, crystals likely adhere to renal epithelial cells, thereby leading to further crystal growth, aggregation and tubular obstruction. Moreover, crystal-cell interactions can lead to an immune and inflammatory response. Cellular changes, including a loss of the epithelial phenotype and the overgrowth of CaOx deposits, can result in CaOx crystal deposition in the interstitial tissue and granuloma formation (Fig. 2.3)1.
Broadly speaking, the pathomechanism of PH at the level of the kidneys can be divided in two initial stages, first CaOx crystallization, and second crystal–cell interactions. Both steps have been investigated as potential drug targets and are described in more detail below.
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
Fig. 2.3: Mechanisms of crystal-induced kidney damage. (1) Elevated urinary oxalate levels in the renal tubules lead to CaOx crystallization. (2) CaOx crystals activate TNF receptors and associated signaling pathways, triggering the expression of crystal binding proteins, such as CD44 and annexin II, on the crystal surface. CaOx crystals can adhere to those cell surface molecules and further grow, aggregate and obstruct the renal tubules. Crystals further trigger epithelial cell injury and inflammation1.
2.3.1 Inhibition of renal CaOx crystallization
Taking a closer look at the first step, developing effective inhibitors of renal CaOx crystallization has been the focus of ongoing research efforts since the 1960s92. Surprisingly little progress has been made in translating findings from mechanistic studies to solid in vivo proofs of concept, and clinical trials61. Conversely, supplementation with urinary alkalizing agents, such as citrate, or potentially CaOx crystallization inhibitory molecules, such as magnesium, in PH patients has been reported to have positive effects93,94. However, clear evidence that the observed effects originate from the inhibition of crystallization is lacking92,95–97. Criticism has been raised on the potency of so-called crystallization inhibitors on renal CaOx crystallization in animal models and humans, and the feasibility of this approach as a therapeutic modality is questioned92,95–97.
A few general rules have emerged over the years, namely the notion that macromolecules tend to be more effective crystal inhibitors than small molecules, and binding to the crystal surface is governed by the negative charge of the compound61. Additionally, in-depth mechanistic studies revealed the influence of subtle changes in the chemical structure of different inhibitors on their CaOx crystal growth modifying effects98–102. For example, comparing citrate with hydroxycitrate, it was found that the insertion/removal of one hydroxyl group can have a drastic impact on the affinity of the molecule to specific crystal faces
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
behavior103. In recent years, the majority of studies have focused on natural urinary inhibitory proteins, such as osteopontin, and synthetic peptidic analogues98–101. A study investigating osteopontin mimetics demonstrated the importance of the number of phosphate groups to achieve close and stable interactions between the inhibitor and the surface of calcium oxalate monohydrate (COM) crystals, the predominant polymorph of CaOx crystals, thereby inhibiting its growth101. A first effort to rationally design and systematically screen peptidic inhibitors was performed by Farmanesh et al.98. Rational design of peptidic inhibitors was based on mimicking common features of natural calcium binding proteins. They therefore varied the number and arrangement of aspartate residues, as calcium binding moieties, and alanine residues acting as spacer groups. Thirteen unique sequences with a total length of 18 amino acids each were designed, differing only in the number and specific sequence of the two amino acids. The peptides were screened for their ability to reduce COM crystallization. It was observed that the efficacy of peptides with the same number of each residue could be increased from 20 to 40% reduction in COM crystallization only by varying their order. This study confirmed the importance of the specific amino acid arrangement and potential steric effects. The proposed platform could enable the synthesis and screening of a large number of peptides, thereby further enhancing the CaOx crystallization inhibitory properties. Moreover, the variation of incorporated amino acids could modulate the synthesized peptides’ physio- chemical properties, such as solubility, which are important determinants for in vivo applications98. Nevertheless, no in vivo data on the efficacy of such peptidic inhibitors are available, which would be needed to better assess the translatability of the approach.
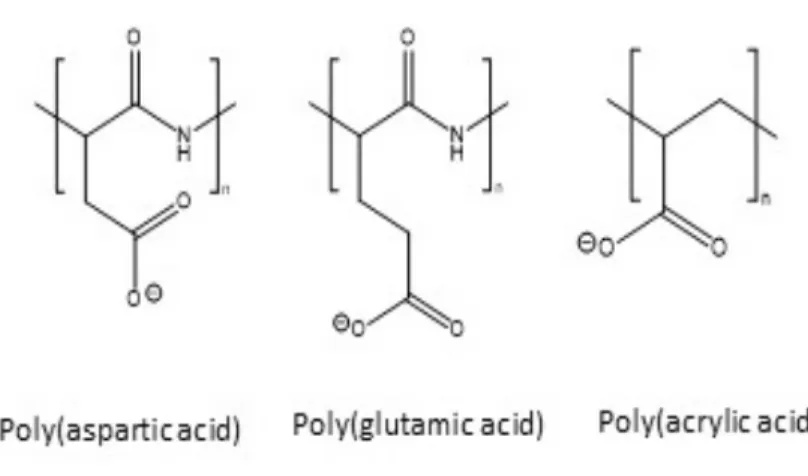
Prior to the work presented in this thesis, to the best of our knowledge, only poly(acrylic acid) showed evidence of inhibition of CaOx crystallization in vitro, as well as in vivo activity in reducing renal CaOx deposition100,104 (Fig. 2.4). In 2004, Jung and coworkers100 demonstrated the superior in vitro efficacy of poly(acrylic acid) (5.1 kDa), a negatively charged non- biodegradable polymer, compared to poly(aspartic acid) (8.3 kDa) and poly(glutamic acid) (6.63 kDa), in inhibiting CaOx crystal growth step velocity in specific directions100. In a later study, it was shown that this polyanion reduced CaOx deposition in rats using a mild model of oxalate nephropathy104. However, no further studies on poly(acrylic acid) were reported104.
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
Fig. 2.4: Polyanions investigated as inhibitors of renal CaOx crystallization. Poly(acrylic acid) showed enhanced potency to reduce CaOx growth rates compared to poly(glutamic acid) and poly(aspartic acid) in vitro, as well as in vivo efficacy100,104.
2.3.2 Blockade of CaOx-induced kidney injury
Increasing understanding of the pathophysiology of crystal associated diseases, and a growing awareness of crystal-induced inflammation contributing to kidney injury are providing cues to potential new treatment options1,105. The NLRP3 inflammasome is of particular interest because of its involvement in a range of crystal-induced diseases, such as gout and atherosclerosis35,36. Its role in mediating renal failure in oxalate nephropathy was shown in the oxalate enriched diet induced hyperoxaluric mouse model described in the previous section. Mice deficient in NLRP3 were completely protected from progressive renal failure caused by the CaOx deposition, thus indicating the molecule’s involvement in mediating the response to CaOx106. It was suggested that the NLRP3/ASC/caspase-1 axis induces IL-1β innate immunity.
Therapeutic blockade with the IL-1β antagonist anakinra reduced CaOx-induced renal injury and neutrophil recruitment in a mouse model of acute oxalate nephropathy, first demonstrating the potential benefit of inflammation inhibitors in CaOx nephropathies37. Specific inhibition of NLRP by the small molecule CP-456,773, similarly reduced intrarenal inflammation and expression of fibrotic markers, and enhanced renal function in mice with chronic CaOx nephropathy107. The NLRP3 inhibitor CP-456,773 might be advantageous over IL-1β antagonists, due to possible oral application, and the potential side effects of broad IL- 1β blockage107. Despite the promising results of NLRP3 pathway inhibition, it should be noted that renal CaOx crystal deposition is not inhibited, thus over prolonged or extensive calcification of the tissue a decline in renal function could occur. Interestingly, TNF receptors have been implicated in mediating CaOx adhesion to renal epithelial cells, potentially regulating the expression of crystal binding proteins, such as CD44 or annexin II, upon CaOx
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
deposition and chronic kidney disease. Small molecule blockade of TNF receptors was tested in the diet induced hyperoxaluric mouse model by preemptive, intraperitoneal treatment with R-7050, a triazoloquinoxaline inhibitor that prevents association of the receptor with intracellular adapter molecules, limiting its internalization and subsequently intracellular TNF receptor signaling responses108. In line with experiments in TNF receptor knockout mice, small molecule blockade of TNF receptorspartially reduced CaOx deposition and tubular injury, as well as improved kidney function compared to vehicle control109.
Taken together, while still in early stages of development, strategies to prevent CaOx-induced kidney damage, comprising both crystallization and inflammation inhibitors, could provide viable treatment options for not only PH, but a broad range of crystal nephropathies. Further research understanding the effects of both types of inhibitors on the pathophysiology together with structure optimizations might pave the way towards the clinical development of effective pharmacological treatments.
2.4 Oxalate degradation in the intestine as a therapeutic modality in PH
Another strategy under investigation for the treatment of PH employs oxalate-metabolizing bacteria. Therein, orally-administered Oxalobacter formigenes (O. formigenes) are utilized to reduce systemic oxalate levels60. O. formigenes, an anaerobic bacterium colonizing the human colon, degrades oxalate as its major source of carbon and energy110. O. formigenes is usually acquired in early childhood and naturally present in the majority of the adult population111,112. However, it was noticed that in recurrent kidney stone formers, as well as enteric hyperoxaluria patients, colonization rates with the bacterium were much lower111,113, indicating its possible involvement in oxalate homeostasis.
Animal studies suggest that O. formigenes colonization favors enteric oxalate elimination, which potentially leads to reduced plasma and ultimately urinary oxalate levels114,115. Intestinal transport of oxalate might result from the diffusion caused by the intestinal degradation of oxalate and the thereby generated transepithelial concentration gradient between the blood and the intestinal lumen, or by specific transporter proteins, such as the anion transporter SLC26A6116,117.
Oral administration of different formulations of O. formigenes, such as a frozen paste or enteric coated capsules, have been tested in clinical trials for the treatment of PH by OxThera.
While the safety of the treatment was confirmed in all of the studies, efficacy data of completed clinical trials on the reduction in urinary oxalate levels as primary endpoint showed variable results to date60,118–120. Limitations might include transient colonization with O.
formigenes, loss of viability during intestinal transit and slow in situ recovery time (i.e. the time for the bacteria to recover their oxalate-degrading capacity from the lyophilized state).
CHAPTER 2: INVESTIGATIONAL THERAPIES FOR PRIMARY HYPEROXALURIA
_____________________________________________________________
treatment additional endpoints, such as plasma oxalate levels, further patient stratification based on baseline kidney function, and prolonged treatment duration are being explored119,121. 24-month interim analysis of an ongoing phase II study in PH1 patients with ESRD pointed out first promising results, showing an approximately 40% reduction in plasma oxalate with O. formigenes treatment122. Further, phase III clinical trials in PH patients with maintained renal function but with a low eGFR and elevated plasma oxalate concentrations are conducted121.
Of note, orally administered oxalate degrading enzymes, namely oxalate decarboxylase, are developed by both OxThera and Allena Pharmaceuticals for the treatment of secondary hyperoxaluria as a first indication123. Secondary or enteric hyperoxaluria describes high urinary oxalate levels acquired by, for example, excess dietary intake of oxalate, malabsorption disorders or ethylene glycol poisoning17. Allena Pharmaceuticals has carried out phase I clinical trials with its lead product candidate reloxaliase, demonstrating reduction in urinary oxalate levels of healthy volunteers on a high oxalate diet in the treatment vs. the placebo control group124. Data of further ongoing or completed trials was made available mid- to end 2019. Promising results were obtained in a phase II study in patients with enteric or primary hyperoxaluria with advanced CKD (stage 4 and ESRD), showing reduction in urinary and/or plasma oxalate levels over weeks 4 to 12. Those results are in line with results of a larger-scale phase III trial in enteric hyperoxaluria patients (no CKD – CKD stage 3), wherein a 23% reduction in 24-hour urinary oxalate excretion compared to baseline levels was achieved125. Currently, the drug is in phase II and III clinical trials for primary and secondary hyperoxaluria respectively126. Further studies are needed to better understand the mechanism of intestinal degradation of oxalate and how it might facilitate the enteric secretion of oxalate. If enzymatic degradation of oxalate can increase the elimination of oxalate similar to O. formigenes, then it might be a viable alternative, with potential advantages in terms of manufacturing and formulation.
2.5 Concluding remarks
PH is a rare, but devastating disease, for which treatment options to date are limited to supportive measures or organ transplantation. In the case of the most severe subtype, PH1, the available treatment modalities can merely delay disease progression towards ESRD10,50. In the last two decades however, the technological advances in novel treatment modalities, such as siRNA127, gene therapy59 or probiotics60, as well as increasing understanding of the PH pathophysiology, especially with regards to crystal-induced kidney injury1, have stimulated the development of promising therapeutic strategies for PH patients. These range from correcting the disease causing mutation in hepatocytes, to preventing kidney damage, and stimulating intestinal breakdown and secretion of oxalate37,59,60,127. siRNAs targeting enzymes involved in the hepatic oxalate synthesis and approaches based on the administration of oxalate metabolizing probiotics treatment are now in an advanced stage with drug candidates