Quantenchemische Untersuchung zur dualen Lumi- neszenz von Zink(II)-Diimin-bis-Thiolat-Komplexen
Quantum Chemical Investigation of the Dual Lumines- cence of Zinc(II)-Diimine-bis-Thiolate Complexes
Bachelorarbeit
von
Frederike Becker
Institut für Theoretische Chemie und Computerchemie Mathematisch-Naturwissenschaftliche Fakultät
Heinrich-Heine Universität Düsseldorf
Erstgutachten: Univ.-Prof. Dr. Christel M. Marian
Eidesstattliche Erklärung
Hiermit erkläre ich, dass die Arbeit „Quantenchemische Untersuchung zur dualen Lu- mineszenz von Zink(II)-Diimin-bis-Thiolat-Komplexen“ von mir eigenständig und nur unter den angegebenen Quellen erstellt worden ist.
Frederike Becker
Danksagung
Zuerst möchte ich Frau Prof. Dr. Christel M. Marian für die Möglichkeit eine Bachelor- arbeit mit interessanter Themenstellung in ihrem Arbeitskreis schreiben zu dürfen, danken. Ebenso danke ich für die hilfreiche Kritik und Anregungen.
Zudem möchte ich Herrn Prof. Dr. Andreas Steffen für die Übernahme der Zweitkor- rektur danken.
Danke auch an Nora Lüdtke für die tatkräftige Unterstützung und Betreuung während der Arbeit und auch bei auftretenden Fragen.
Außerdem möchte ich dem Arbeitskreis für die Hilfe und die Unterstützung danken.
Kurzfassung
In der vorliegenden Arbeit werden verschiedene Konformere des (4-Thiophe- nol)
2Zink(1,10-Phenanthrolin) Komplexes untersucht. Ziel dieser Untersuchung ist es, die Konformere quantenchemisch zu charakterisieren und mit vorhandenen Literatur- daten zu vergleichen.
Das Ergebnis der Untersuchung ist, dass die Absorptionen und Geometrieparameter gut mit der Literatur übereinstimmen. Die Konformere untereinander sind sich ähn- lich, bis auf die Lage der Thiophenolringe.
Bei der Emission werden deutliche Unterschiede zu der Literatur gefunden. Die Maxi- ma sind um ca. 1000 nm verschoben. Dabei liegt das Maximum des berechneten Komplexes nicht im sichtbaren Bereich.
Abstract
In the following thesis different conformers of (1,10-Phenanthroline)bis(4-toluenethio- lato)zinc(II) complex are investigated. Aim of the investigation is to characterize quantenchemical the conformers and to compare the results with the literature. This is performed on the basis of actually quantum chemistry methods.
Results of the investigation are that the absorptions and geometry parameters com- port with the literature. The conformers are similar among themselves, except the po- sition of the toluenthiolato-rings.
The emission are obvious different to the literature. Maxima are shifted of 1000 nm.
At that the maximum of the computed complexes are not in the visible area.
Inhaltsverzeichnis
Abkürzungsverzeichnis ……….………….. iv
1. Einleitung ...…...…...…...…..…...…...…….. 1
1.1. Stand der Forschung ....…...…...………...……...… 3
2. Theorie ....………...…...………...……...…..……...…...…. 5
2.1. Lumineszenz ...………...…...……...…...…. 5
2.2. Anregungsarten ...…...……...…...…...….…....….………..…. 6
2.3. Franck-Condon-Spektrum ...…...…...…...…..…. 8
3. Quantenchemische Methoden ...……...…...…..…....………... 8
3.1. Dichtefunktionaltheorie DFT ...…..…...…...…….….... 8
3.2. Zeitabhängige DFT TDDFT ...…...……...…...………... 9
3.3. Dispersionskorrektur ...…...…...…...…...…...….. 10
3.4. Multireferenz-Konfigurationswechselwirkung MRCI ...…...……...…. 11
3.5. DFT/MRCI ...…...……...…...…...…...….…….. 11
3.6. R2018 Hamiltonian ...…....…...…….……...……...…...…. 12
4. Programme ...…...……...…...…...…...…...……….. 12
5. Technische Parameter zu den Rechnungen ...……….…..…...…. 13
5.1. Basissätze ...…...……...…..….………...…. 13
5.2. Effektives Rumpfpotenzial ...…...…...……...…...……....…. 13
5.3. verwendete Funktionale ...…...……...…...………….…….... 14
6. Ergebnisse und Diskussion ...…....…...…………...…...…..…. 15
6.1. Vergleich der Konformere im Grundzustand ...….…...…...…. 16
6.2. Vergleich der Konformere im ersten angeregten Triplettzustand (LLCT)19 6.3. Vergleich der Konformere im ersten angeregten Singulettzustand …. 24 6.4. Anregungen und Emissionen.…...…....…...…....…....………... 27
7. Zusammenfassung ..…..………...………...………..…. 36
8. Literaturverzeichnis ..…...…...…...……….... 37 9. Anhang ..…...…...…...…...……….. I
9.1. Tabellen ..…...…...…...……….. I
9.2. Abbildungen ..…...…...…...…...…...……. LI
Abkürzungsverzeichnis
OLED: organic light emitting device
TADF: thermally activated delayed fluorescence LLCT: Ligand-zu-Ligand-Charge-Transfer
IC: internal conversion ISC: intersystem crossing
rISC: reverse intersystem crossing
MLCT: Metall-zu-Ligand-Charge-Transfer LMCT: Ligand-zu-Metall-Charge-Transfer CT: Charge-Transfer
LC: ligandenzentrierter Übergang DFT: density functional theory
TDDFT: time-dependent density functional theory
TDDFT-TDA: time-dependent density functional theory-tamm-dancoff approximation MRCI: multireference configuration interaction
MRCISD: multireference configuration interaction singles and doubles
ECP: effective core potential
1. Einleitung
Licht-emittierende Metallkomplexe haben in den letzten Jahren deutlich an Bedeu- tung für fluoreszierende und phosphoreszierende Elemente gewonnen. Zu diesen zählen organische Leuchtdioden (organic light emitting device OLED) für Bildschirme in Handys oder Fernsehern. [1]
OLEDs sind aus verschiedenen Schichten aufgebaut. Durch Anlegen von Spannung werden Überschusselektronen und Löcher erzeugt. Die Schichten bestehen aus dün- nen organischen Filmen, durch welche die Elektronen und Löcher migrieren können.
Rekombinieren Elektronen mit Löchern, können Exitonen entstehen. Ein Exiton kann zu 25% ein Singulettcharakter und zu 75% einen Triplettcharakter haben. Die Grund- lage der Aufteilung der Ausbeute ist, dass sich der Triplettzustand in drei Zustände aufteilt, wohingegen der Singulettzustand auf nur einem Zustand aufbaut. [1-3]
Es gibt drei Generationen von OLEDs. Die erste Generation sind fluoreszierende OLEDs, die zweite Generation phosphoreszierende OLEDs und die dritte Generation basiert auf thermisch aktivierter verzögerter Fluoreszenz (TADF).
Die erste Generation von OLEDs basieren auf Komplexen, welche eine prompte Flu- oreszenz aufweisen. Der Vorteil von dieser Generation ist, dass die Farben deutlich sind und die Lebensdauer kurz ist. Jedoch beträgt die innere Quantenausbeute nur 25% beträgt.
Die zweite Generation von OLEDs wird der ersten Generation vorgezogen. Da ein ef-
fizienter Phosphoreszenzemitter sowohl die Strahlung vom Singulett- als auch vom
Triplettzustand aufnehmen kann. So können bis zu 100% der angeregten Zustände
erfasst werden. [1] Die meisten phosphoreszenten OLEDs arbeiten heutzutage mit
Komplexen, bei denen das Komplexmetall eine hohe Kernladungszahl hat und somit
eine höhere Masse besitzt. Als Komplexmetalle werden aktuell Ruthenium(II), Iridi-
um(III), Platin(II), Rhenium(I) oder Gold(III) verwendet. Dieses ist für die benötigte
große Spin-Bahn-Kopplung, welche für ein intersystem crossing (ISC) benötigt wird,
wichtig. Ein Intersystem Crossing ist von hoher Bedeutung, da dieser Prozess für
den Übergang in den T
1-Zustand notwendig ist. Der Prozess ist wegen der Spinum-
kehr spinverboten, kann aber aufgrund von genügend großen Spin-Bahn-Kopplun-
gen trotzdem stattfinden. [4]
Für die dritte Generation OLEDs werden TADF Emitter benötigt. Damit im Gegensatz zu der ersten Generation eine große innere Quantenausbeute mit der Fluoreszenz erreicht werden kann, werden Triplett-Zustände, die wieder in einen angeregten Sin- gulett-Zustand übergehen, miteinbezogen. So kann eine zeitlich verzögerte Fluores- zenz beobachtet werden. Dazu wird ein reverse intersystem crossying (rISC) vom T
1in den S
1benötigt. Ein Molekül zeigt einen solchen rISC, wenn T
1und S
1mit einer Energielücke von ΔE
ST= ≤ 0,1 eV auseinander liegen. Dieses Phänomen tritt bei in- tramolekularen Charge-Transfer Übergängen innerhalb von Systemen auf, die räum- lich getrennte Donor- und Akzeptoranteile beinhalten. [5] Die Vorteile dieser Genera- tion sind eine Kombination aus den beiden Generationen davor. Die Lumineszenz hat eine kurze Lebensdauer, kann aber auch eine hohe innere Quantenausbeute auf- weisen. Nachteilig ist, dass TADF-Emitter eine niedrige Strahlenratenkonstante besit- zen. [6]
Für solche TADF-Emitter eignen sich einige Platin(II)-, Iridium(II)- und Kupfer(I)-Kom- plexe. Kupfer(I)-Komplexe haben eine kurze Lebenszeit von ca. 10 µs und sind damit genauso kurzlebig wie Platin(II)- oder Iridium(II)-basierende Emitter. Der Vorteil von Kupfer gegenüber den beiden anderen Emittern ist, dass es sich um ein d
10-Elemet handelt. Dadurch werden die problematischen metallzentrierten d-d* Übergänge ver- mieden. Diese Übergänge sind strahlungslos und konkurrieren zu den strahlungsrei- chen Prozessen. [7]
Nach dieser Feststellung werden auch andere d
10-Komplexe untersucht, ob sich die- se als Emitter eignen. Eine weitere Komplexgruppe im Bereich der d
10-Elemente sind Zink(II)-Komplexe. In der Literatur werden einige vermutlich phosphoreszierende Zink(II)-Komplexe genannt. Das Problem bei Zink ist aktuell, dass der Großteil der Zink(II)-Komplexe nur fluoresziert. Daher wird jetzt nach Zink(II)-Komplexen gesucht, welche in den Triplett-Zustand angeregt werden können, und sich somit als phospho- reszente oder TADF Emitter eignen. [8]
Einer dieser vermutlich phosphoreszierenden Zink(II)-Komplexen ist das (4-Methyl-
Thiophenol)
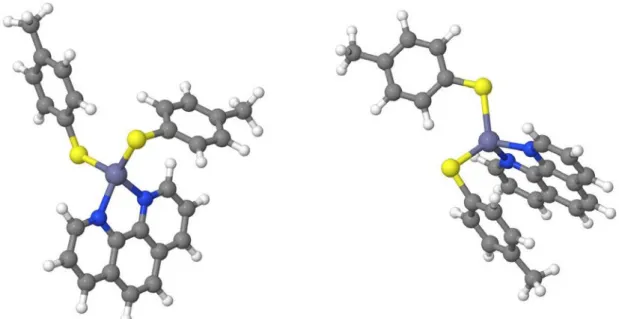
2Zink(1,10-Phenanthrolin). Dieser Komplex (Abb. 1) wird in der vorliegen-
den Arbeit unter Berücksichtigung der Lumineszenzeigenschaften untersucht. Es
reszierender Zink(II)-Komplexe gefunden werden. Ebenso sollen für die Suche neu- artiger phosphoreszierender oder TADF-fähiger Zink(II)-Komplexe Informationen ge- sammelt werden.
Der behandelte Komplex ist aus zwei Arylthiolatliganden und einem a-Diiminligand aufgebaut.
Abb. 1: Zn(II)(4-CH3-C6H4S)2(1,10-Phenanthrolin)
1.1. Stand der Forschung
Der Komplex wurde experimentell von Highland et. al. untersucht [9]. Dabei wurde festgestellt, dass der Komplex einen multiexponentiellen Zerfall der Lumineszenz aufweist. In einem Kristall werden Lebensdauern zum einen von t = < 15 ns, von t = 7 ms, sowie von t = 0,34 s bei einer Temperatur von 6,5 K gemessen. Im Vergleich dazu werden in einem starren Glas aus Isobutyronitril bei einer Temperatur von 77 K zwei Lebensdauern gemessen. Einmal von t = 0,6 s mit einer Emissionsquantenaus- beute von 0,07 bei der lokalen Anregung auf dem 1,10-Phenanthrolin und eine zwei- te mit t = 8,3 µs mit einer Emissionsquantenausbeute von 0,03 bei einem Ligand-zu- Ligand-Charge-Transfer. Es handelt sich hierbei ebenfalls um experiementelle Daten.
[8]
Außerdem wurden zu den Lebensdauern in dem Komplex Emissionsspektren in Lö- sung [8,10] sowie in einem starren Glas [11] bei 77 K aufgenommen. Diese zeigen ei- nen ähnlichen Verlauf.
Bei der Untersuchung im Kristall ist aufgefallen, dass die Intensität des langlebigeren Teils mit steigender Gittertemperatur sinkt, wohingegen die Intensität des kurzlebige- ren Teils mit steigender Gittertemperatur steigt. Allerdings verschwindet die kurzlebi- ge Komponente auch bei geringen Temperaturen nicht. Die langlebige Komponente ist ab ca. 35 K für die Experimentatoren nicht mehr sichtbar gewesen. Im starren
N N
Zn S S
Glas lassen sich sowohl die Lumineszenzen aus der lokale Anregung, als auch der des Ligand-zu-Ligand-Charge-Transfer noch bei 77 K beobachten.
Beobachtet wurde, dass die langlebigste Lumineszenz im blau-grünen Spektralbe- reich emittiert. Die beiden kurzlebigeren Lumineszenzen emittieren dahingegen im gelben und roten Spektralbereich.
Die Experimentatoren schlagen vor, dass die langlebigste Lumineszenz aus einer
3ππ* Anregung des 1,10-Phenanthrons resultiert. Im Gegensatz dazu strahlen die
beiden kurzlebigeren Lumineszenzen aus einem Ligand-zu-Ligand-Charge-Transfer (LLCT) Zustand. In dem vorgeschlagenen Energiemodell nehmen die Experimentato- ren aufgrund der oben genannten Temperaturabhängigkeit eine Energiebarriere an, die überwunden werden soll. (Abb. 2) [9]
Abb. 2: Vorgeschlagenes Energiemodell der Experimentatoren
Ebenso sagen die Experimentatoren, dass dieser Komplex gegen Kasha‘s Regel für organische Verbindungen und gegen die Regel von Demas und Crosby für die Lumi- neszenz von Übergangsmetallkomplexen für offenschalige d-Systeme verstößt.
Die Regel von Kasha besagt, dass die Emission nur aus dem niedrigsten Zustand ei- ner Multiplizität stammt. Dieses stimmt nicht mit dem Ergebnis der Experimentatoren überein. [9]
Zudem besagt die Regel von Demas und Crosby, dass die Emission in einem Über-
gangsmetallkomplex mit offenschalige d-Systeme aus dem energetisch niedrigsten
angeregten Zustand oder aus dem angeregten Zustand herrührt, welcher eine signifi-
Wegen dieser Verstöße sollte der untersuchte Komplex entsprechend unter Berück- sichtigung der besonderen Charakteristik der geschlossenen d-Schale untersucht werden. [9]
2. Theorie
2.1. Lumineszenz
Die Lumineszenz ist ein Überbegriff für zwei Strahlungsarten, welche mit dem Ja- blonski Diagramm (Abb. 3) veranschaulicht werden können. Zum einen gibt es die Fluoreszenz und zum anderen die Phosphoreszenz. Bei beiden Strahlungsarten han- delt es such um eine strahlende Relaxation vom angeregten Singulett- (Fluoreszenz) / Triplett-Zustand (Phosphoreszenz) in den Grundzustand. Die prompte Fluoreszenz wird spontan nach Anregung emittiert. Dieses geschieht einige Nanosekunden nach Abschalten der Anregung. Diese Emission ist kurzlebig. Im Gegensatz dazu ist die Phosphoreszenz langlebig, das heißt die Strahlung kann meist für Sekundenbruch- teile nach der Abschaltung der Anregung emittiert werden. [13]
Die prompte Fluoreszenz wird durch einen strahlungsreichen S
1– S
0Übergang ge- kennzeichnet. Neben der Fluoreszenz gibt es zwei strahlungslose Konkurrenzpro- zesse aus dem S
1-Zustand. Zum einen das internal conversion (IC). Dieser Prozess ist ein Übergang zwischen verschieden Zuständen der gleichen Multiplizität und strahlungslos. Zum anderen das intersystem crossing (ISC). Dieses ist ein strah- lungsloser Übergang zwischen verschiedenen Zuständen unterschiedlicher Multiplizi- tät. Zum Beispiel zwischen S
1und T
1. Die Fluoreszenz überwiegt, wenn der strah- lungsreiche Prozess schneller verläuft als die strahlungslosen. [14]
Die Phosphoreszenz ist ein strahlungsreicher Übergang von einem Triplett-Zustand in den Grundzustand. Aufgrund der Umkehrung des Spins ändert sich die Multiplizi- tät. Als Konkurrenzprozess zu der Phosphoreszenz gibt es das strahlungslose ISC.
ISC ist spinverboten, kann aber aufgrund großer Spin-Bahn-Wechselwirkungen trotz-
dem stattfinden. [14] Aufgrund der Spinumkehrung ist die Phosphoreszenz langlebi-
ger als die prompte Fluoreszenz.
Abb. 3: Jablonski Diagramm [15]
2.2. Anregungsarten
In Übergangsmetallkomplexen gibt es verschiedene Arten von Anregungen. Zum ei- nen gibt es Übergänge, die innerhalb eines Liganden (ligandenzentriert LC) stattfin- den und solche, die ligandenübergreifend (Ligand-zu-Ligand-Charge-Transfer LLCT) sind. Bei diesen Anregungen sind die Elektronen des Metalls nicht beteiligt. Es gibt bei anderen metallorganischen Komplexen oft Anregungsarten, bei denen das Metall involviert ist. Diese nennen sich Metall-zu-Ligand-Charge-Transfer (MLCT), Ligand- zu-Metall-Charge-Transfer (LMCT), sowie metallzentrierte (MC) Übergänge. Von die- sen drei Übergängen denken die Experimentatoren, dass diese nicht in dem vorlie- gendem Komplex zu einer Anregung beitragen. [9,11] In OLEDs sind besonders MC Übergänge zu vermeiden, da diese strahlungslos sind und mit den strahlungsreichen Prozessen konkurrieren. In d
10-Metallkomplexen gibt es keine MC Übergänge, da diese eine geschlossene d-Schale besitzen. [7]
Es wird immer der beteiligte Teil der Anregung zuerst genannt aus dem die Elektro-
nen angeregt werden. Als zweites wird der Teil genannt, in den die Elektronen ange-
regt werden. Bei den Übergängen innerhalb des Liganden werden von einem besetz-
ten Orbital in ein unbesetztes Orbital des Liganden Elektronen übertragen. Bei dem
ligandenübergreifenden Übergang, auch Charge-Transfer (CT) genannt, wird aus
dem Donorliganden ein Elektron aus einem besetzten in ein unbesetztes Orbital des
Akzeptorliganden geliefert. In einem verbrücktem Ligand kann es auch zu einem CT
LLCT
Der Interligandaustausch, auch Ligand-zu-Ligand-Charge-Transfer (LLCT) genannt, benötigt eine
π-/ n-Donor- und einen π-Akzeptorliganden. Der
π/n-Donor ist in dem behandelten Komplex das 4-Methythiophenol. Der π-Akzeptorligand ist durch das 1,10-Phenanthrolin gegeben. [4]
ππ*
Der ππ* Übergang ist ein ligandenzentrierter (LC) Übergang. [17]
Bei diesem Übergang sind Donor- und Akzeptoranteil einem Liganden zugeordnet und nicht räumlich getrennt. [18]
MC
Hierbei handelt es sich um eine d-d* Anregung. Das bedeutet, dass dabei aus einem besetzten Orbital des Metalls in ein unbesetztes Orbital des Metalls angeregt wird.
Diese Art der Anregung kann bei Metallen vorkommen, welche keine d
10-Konfigurati- on haben. Die Emission aus dieser Anregung ist strahlungslos. [7]
MLCT
Bei einem MLCT Übergang wird ein Elektron aus einem d Orbital des Metalls in ein unbesetztes antibindendes Orbital des Liganden übertragen. Das Metall wird oxidiert und der beteiligte Ligand wird reduziert. Der Metallkomplex besitzt zudem ein gerin- ges Oxidationspotential. [18]
LMCT
Bei diesem Übergang wird von einem besetzten Orbital eines Liganden ein Elektron
in ein unbesetztes Orbital des Metalls angeregt. Hierbei hat das Metall ein hohes
Oxidationspotential. [18]
2.3. Franck-Condon-Spektrum
Das Franck-Condon Prinzip bestimmt Wahrscheinlichkeiten des Übergangs zwischen verschiedenen elektronischen Zuständen. Diese Annahme fußt darauf, dass die Elektronen aufgrund der geringeren Masse gegenüber den Kernen kaum nennens- werten Einfluss auf diese haben. Aufgrund dessen ändert sich bei dem Wechsel von elektronischen Zuständen der Kernabstand nicht und die Anregung erfolgt vertikal.
Bei der Absorption wird mit höchster Wahrscheinlichkeit in den Zustand angeregt, welcher in der Potentialdarstellung mit dem Maximum seiner Wellenfunktion senk- recht über dem Maximum des Schwingungsgrundzustandes liegt. [19]
Mittels des Franck-Condon Prinzips werden diskrete Spektren berechnet, welche auf Grundlage der Übergänge zwischen zwei stabilen Zuständen erstellt werden. Be- rechnet werden dabei die Übergänge zwischen den vibronischen Zuständen aus dem angeregten Zustand und dem Grundzustand. Es wird die Intensität gegen die Wellenlänge aufgetragen. [19]
3. Quantenchemische Methoden 3.1. Dichtefunktionaltheorie DFT
Die Dichtefunktionaltheorie (DFT) ist eine weit verbreitete Möglichkeit, um mit relativ wenig Rechenaufwand Moleküle zu berechnen. Im einfachsten Fall kann die Energie über die Elektronendichte berechnet werden, dafür wird die Wellenfunktion nicht be- nötigt. Da allerdings das exakte Funktional der Energie nicht bekannt ist, müssen Nä- herungen gemacht werden. Aus diesem Grund führten Kohn und Sham Orbitale in die Rechnung mit ein. Dadurch wird die Berechnung wieder abhängig von der Wel- lenfunktion und aufwendiger. Allerdings ist diese Methode weniger rechenintensiv als andere auf wellenfunktionsbasierende Berechnungen und liefert bessere Ergebnisse als die Hartree-Fock Näherung. Denn bei der Hartree-Fock Näherung werden keine Korrelationsanteile berechnet. [20,21]
Bei der Kohn-Sham-DFT wird ein fiktives Referenzsystem aus so vielen nicht-wech-
selwirkenden Elektronen eingefügt, wie es wechselwirkende Elektronen in dem Sys-
wie das originale System. Bei dem fiktiven System kann die exakte Grundzustands- dichte ausgerechnet werden. Aus dieser Dichte, stammend aus den Orbitalen, nicht von der Elektronendichte, kann die kinetische Energie genau berechnet werden. Die- se kinetische Energie kann mit einem genäherten Korrekturterm zur genäherten kine- tischen Energie des originalen Systems führen. Die kinetische Energie ist ein Teil des Austauschkorrelationsfunktionals. Das Austauschkorrelationsfunktional fasst alle Ter- me zusammen, die nicht genau ausgerechnet werden können. Dazu zählen Cou- lomb-, Austausch- und Korrelationsanteile, sowie die kinetische Energie. [20,21]
3.2. Zeitabhängige DFT TDDFT
Die zeitabhängige DFT (TDDFT) ist die meist benutze Näherung für die Berechnung der Eigenschaften angeregter Zustände von mittleren bis großen Molekülen. Zum Beispiel für die Geometrie angeregter Zustände, Oszillatorstärken oder Anregungs- energien. Grundlage für die Theorie ist das Runge-Gross-Theorem.
Die Eigenwertgleichung für die TDDFT lautet: [22]
[
BA*
AB* ][YX]
=ω[ 1 0 - 1 0 ][
XY] (2)
Aia , jb=δijδab(ϵa−ϵi)+(ia∣jb)+(ia∣fxc∣jb)
Bia , jb=(ia∣bj)+(ja∣fxc∣bj)
(3)
In der Gleichung (2) steht für die Anregungsenergien und f
xcfür den Austauschope- rator.
e ist die Orbitalenergie für die Ein-Elektronenorbitale. Dabei beschreiben die Indizes
a und b besetzte und i und j unbesetzte Orbitale. [22] Die Nomenklatur (ia|jb) ist in der Mulliken Notation für Zweielektronenintegrale geschrieben.
Große Probleme hat die TDDFT zum Beispiel bei der Beschreibung von doppelt an- geregten Zuständen.
Ebenso kann die TDDFT nicht die Charge-Transfer-Zustände korrekt beschreiben.
Hierbei kann der Betrag des Fehlers um mehrere Elektronenvolt von dem richtigen
Ergebnis abweichen. Durch gute Näherung des Austauschintegrals kann bei Charge- Transfer-Zuständen das richtige asymptotische Verhalten (1/R) hervorgerufen wer- den. Dieses ist der Fall, wenn sie Singularitäten aufweisen. Singularitäten treten auf, sobald die Überlappung des elektronendonierenden und des elektronenakzeptieren- den Orbitals genähert gleich Null ergeben. [22]
TDDFT-TDA ist eine weitere Näherung zu der TDDFT. Die Tamm-Dancoff Näherung (TDA) vernachlässigt die Bedeutung das B-Matrixenelement aus Gleichung (2). [22]
AX=ω X
(4)
Durch die Vernachlässigung des B-Matrixelements wird der Rechenaufwand erneut geringer, wohingegen das Ergebnis nah an dem der TDDFT-Rechnung liegt. Einen großer Unterschied zwischen TDDFT und TDDFT-TDA ergibt sich bei der Berech- nung von Molekülen, welche ein triplettnahes Instabilitätsproblem haben. Bei der TDDFT-TDA wird die Anregungsenergie nicht so systematisch unterschätzt. Aus die- sen Gründen ist die TDDFT-TDA Rechnung eine nutzbare Alternative zu der TDDFT- Rechnung. [23]
3.3. Dispersionskorrektur
Die Dispersionskorrektur schließt die London-Dispersionswechselwirkungen mit ein.
Denn diese Wechselwirkung wird bei der DFT nicht berücksichtigt. Diese ist jedoch
relevant, da die Wechselwirkung selbst bei Atomen auftritt und sich bei Molekülen
noch mehr auf die Energie auswirkt. Ohne die Berücksichtigung der Dispersion hat
die DFT Rechnung eine hohe absolute Abweichung. Mit Dispersionskorrektur ist die
Abweichung nicht so groß. Deshalb wurde nachträglich bei den DFT Rechnungen
eine Dispersionskorrektur angewendet. Verwendet wird die DFT-D3-Korrektur. [24]
3.4. Multireferenz-Konfigurationswechselwirkung MRCI
Die Multireferenz-Konfigurationswechselwirkung (MRCI) ist eine korrelierte lineare Variationsmethode für den Grundzustand und die elektronisch angeregten Zustände.
Dabei handelt es sich um eine ab initio Methode, bei welcher durch eine Linearkom- bination von Slater-Determinanten, die verschieden substituiert sind, Berechnungen durchgeführt werden.
Bei der MRCISD werden einfach- und doppeltsubstituierte Determinanten verwendet.
Würden alle höheren Anregungen berücksichtigt werden, würde ein full CI erhalten werden, welche die exakte Korrelationsenergie wiedergibt.
Die Rechnung kann vereinfacht werden, indem einige unbesetzte und einige besetz- te Orbitale eingefroren werden. Diese sind somit nicht an der Anregung beteiligt. Bei dem kleinstmöglichen Fall der Einfrierung sind nur HOMO und LUMO nicht eingefro- ren. Das muss aber nicht zwangsläufig durchgeführt werden, da auch oft gar keine Orbitale eingefroren werden. [20,25]
3.5. DFT/MRCI
Die DFT/MRCI ist eine Kombination aus der Dichtefunktionaltheorie (DFT) und der Multireferenz-Konfigurationswechelwirkung (MRCI). Dabei werden die beiden Metho- den so kombiniert, sodass das beste aus jeder Methode verwendet wird. Bei der Me- thode handelt es sich um eine empirische Näherung.
Kombiniert werden die beiden Methoden, da sie unterschiedliche Teile der Korrelati- on unterschiedlich gut beschreiben. Zum einen beschreibt die MRCI die statische Korrelation, welche mit der DFT nicht berechnet wird. Die DFT beschreibt die dyna- mische Korrelation. Die MRCI berechnet ebenso die dynamische Korrelation, aller- dings ist die Berechnung der dynamischen Korrelation mittels MRCI langwieriger.
Aus diesem Grund werden durch die Skalierung einiger Außerdiagonalelemente die dynamische Korrelation bei der MRCI herausgefiltert.
Für die Skalierung werden einige an experimentelle Daten angepasste Parameter
eingeführt, welche nicht elementspezifisch, sondern funktionalspezifisch sind. Aktuell
gibt es nur für das BH-LYP Funktional Parametersätze.
Diese Methode eignet sich auch, im Gegensatz zu TDDFT, für angeregte Zustände mit Doppelanregung und Charge-Transfer-Zuständen. Die Kombination der Metho- den ist genauer als TDDFT. [25-28]
3.6. R2018 Hamiltonian
Der originale Hamilton-Operator wurde von Grimme und Waletzke entwickelt. Dieser Hamilton-Operator weist allerdings bei der Beschreibung von Multichromophorsyste- men Probleme auf. Eine Weiterentwicklung dieses Operators ist der R2016 von Lys- kov et al. (R2016). Dieser behebt das Problem von Multichromophorsystemen für or- ganische Moleküle mit lokal angeregte Singulett- und Triplett-Zustände. Jedoch ist dieser Hamilton-Operator nicht für Systeme mit ungeraden Elektronenanzahl geeig- net. Deshalb gibt es eine Weiterentwicklung, den R2017 von Heil et al. Darauf auf- bauend wurde der R2018 Hamilton-Operator, welcher sich auch für Übergangsme- tallkomplexe eignet, entwickelt. In dem R2018 Hamilton-Operator wurde die Dämp- fungsfunktion dahingehend verändert, dass diese nicht so früh, dafür steiler, abfällt.
Dadurch werden die Anregungsenergien, auch für Charge-Transfer Zustände, genau- er beschrieben, unabhängig von der Multiplizität. [29]
4. Programme
Zum Erstellen der Konformere des Komplexes wurde das Programm Avogadro Versi- on 1.2.0 [30] verwendet.
Zur Geometrieoptimierung wurde das Programm Turbomole Version 7.0 [31] verwen- det. Dabei wurde die Schwingungsfrequanzanalyse mit dem Programm snf [32]
durchgeführt.
Bei der DFT/MRCI-Rechung wurde der CI-Referenzraum auf 8 Elektronen in 8 Orbi-
tale mit Doppelanregung begrenzt. Die untersten 42 Orbitale wurden mit dem Pro-
gramm rimp2prep eingefroren, da diese kaum Einfluss auf die berechneten Zustände
haben.
berechnet. Als Hamilton-Operator wurde der R2018 Hamilton-Operator [29] verwen- det.
Die Franck-Condon-Spektren wurden mit VIBES [33] Version 1.2 berechnet.
Die Visualisierung und Vermessung wurde mit Jmol Version 14.4.4_2016.04.14 [34]
durchgeführt.
5. Technische Parameter zu den Rechnungen 5.1. Basissätze
Basissätze dienen zur genäherten Berechnung von Atomen. Es handelt sich um kon- trahierte Gaußfunktionen. Mit der einfachste Basissatz ist der split valence (SV) Typ.
Dieser wird in der polarisierten Form (SV(P)) für die leichten Elemente, wie Wasser- stoff, Kohlenstoff und Stickstoff, genutzt. Die Vorsilbe „def“ steht für „default“ „Stan- dard“.
Für Schwefel wird die Basis def2-SVPD verwendet, welche eine neue Serie der Stan- dardbasen (def2) darstellt. [35,36]
Für Zink wurde der Basissatz 10-mdf 6s5p3d benutzt. [37]
Für die DFT/MRCI Rechnung wird für alle Atome eine Auxiliarbasis genutzt.
Für Zink wird als Auxiliarbasis der Basissatz TZVP (triple zeta valence plus polarizati- on) verwendet. [38]
5.2. Effektives Rumpfpotenzial
Bei schwereren Atomen, wie zum Beispiel Zink, würde die Berechnung für alle Elek- tronen lange Rechenzeiten und Speicherkapazitäten einnehmen. Damit die Rechen- zeiten verkürzt werden, werden die Rumpfelektronen eingefroren. Deswegen werden sie durch ein Pseudopotential ersetzt, welche den ab initio Charakter beibehalten.
Das Pseudopotential wird in dem Hamilton-Operator als Einelektronenanteil wieder- gefunden.
Bei diesem Komplex wurde für das Zn defpp-ecp als effektives Rumpfpotenzial ver-
wendet, das anhand einer relativistischen Atomrechnung parametrisiert wurde. [20]
5.3. verwendete Funktionale PBE0
Das PBE0 Funktional, entwickelt von Perdew, Burke und Ernzerhof, ist ein parame- terfreies Hybridfunktional und setzt sich aus drei Viertel des PBE Austausches und einem Viertel des Fock Austausches zusammen. Die Elektronenkorrelation wird vom PBE Funktional beigesteuert. Durch den fixen Beitrag des Fock Austauschterms wird der Fehler der Selbstwechselwirkung reduziert. [39]
EPBExc 0=
1 4
Ex+3
4
EPBEx +EcPBE(5)
BH-LYP
Hierbei handelt es sich ebenfalls um ein Hybridfunktional. Für die Austausch-Wech- selwirkung wird eine Hälfte exakt aus der Hartree-Fock-Theorie und die andere Hälf- te über die lokale Dichtenäherung, beziehungsweise durch das B88 Funktional ermit- telt. [26]
Die Korrelation wird mithilfe des LYP Funktionals beschrieben. [40]
EBHLYPxc =
0,5
EHFx +0,5
ELDA/Bx 88+EcLYP(6)
6. Ergebnisse und Diskussion
Der Komplex (4-Methyl-Thiophenol)
2Zink(1,10-Phenanthrolin) kann verschiedene Konformere aufweisen. Zwei dieser Konformere wurden untersucht. Konformer A ist selbsterstellt mit Avogadro, Konformer B basiert auf Kristalldaten aus der Literatur [41]. Diese beiden Konformere wurden miteinander und mit der Literatur [41] vergli- chen. Die Literaturdaten [41] wurden mittels Röntgenstrukturanalyse bei 77 K erstellt.
Andere Literaturdaten [42] wurden bei 298 K mittels Röntgenstrukturanalyse erstellt, weichen aber zum Teil stark von Standardbindungslängen im 1,10-Phenanthrolin ab, weshalb dazu kein Vergleich gezogen wurde. Die Daten befinden sich im Anhang in den Tabellen A7 und A8.
Bei dem Literaturvergleich werden die Bindungslängen und -winkel zu den Wasser- stoffatomen nicht mit berücksichtigt, da diese mittels Röntgenstrukturanalyse nicht genau erfasst werden. Ebenso wurden keine Diederwinkel gemessen.
In Abbildung 4 ist die Nummerierung des Komplexes dargestellt, die für den Geome- trievergleich relevant sind.
Abb. 4: Zweidimensionale Darstellung mit Nummerierung der Atome, der grüne 4-Methyl- Thiophenolligand ist unter das 1,10-Phenanthrolin gedreht, der blaue 4-Methyl-
Thiophenolligand dreht sich von dem 1,10-Phenanthrolin weg.
Die Konformere wurden zuerst mit PBE0 optimiert. Dort waren die LLCT Übergänge bei der Anregung energetisch tiefer abgesenkt als die LC Übergänge. Aus diesem Grund wurden die Konformere mit BH-LYP erneut optimiert. Es wurde mit BH-LYP
N N
Zn S S
H H
H H
H H H H H
H H
H H
H H H H
H
H HH H
5 4 6
8 7 11
12 10
25 13 9
14 15
23 22
16
1
2
53 24
48 42
26 3 27
18
17
21 20
47 52 49
44
41 40
43 46
28 36
37
29 3 3 35
30 31
32 34
39 38
45
50 19 51
optimiert, da in diesem Funktional der Fock-Anteil größer ist, als in dem Funktional PBE0. Dadurch wird der Fehler der Selbstwechselwirkung reduziert. Allerdings gibt das PEB0-Funktional gute Ergebnisse für Übergangsmetalle. [39]
Damit die LC Übergänge energetisch in eine bessere Lage gerückt werden, wurde die Geometrie des Komplexes verändert. Dazu wurde zuerst von dem 1,10-Phenan- throlin der erste angeregte Triplett-Zustand optimiert. Danach wurde von dieser Struktur die Grundzustandsgeometrie der Konformere dahingehend geändert, dass die Bindungslängen und -winkel, sowie die Diederwinkel des 1,10-Phenanthrolins dem der optimierten T
1-Geometrie entsprechen. Die anderen Parameter des Komple- xes blieben in der Grundzustandsgeometrie. Dieser Vorgang wurde an beiden Kon- formeren durchgeführt.
6.1. Vergleich der Konformere im Grundzustand
Abb. 5: Darstellung der optimierten Grundzustandsgeometrie mit PBE0 zweier Konformere des Zn(II)(4-CH3-C6H4S)2(1,10-Phenanthrolin), links Konformer A, rechts Konformer B, die Schwefelatome werden gelb abgebildet, die Wasserstoffatome weiß, die Kohlenstoffe grau,
die Stickstoffe blau und das Zinkatom lila.
Bei dem Vergleich der Grundzustandsenergien der beiden Konformere A und B, wird
deutlich, dass das Konformer B energetisch stabiler ist. Dieses ist für beide Funktio-
nale gleich.
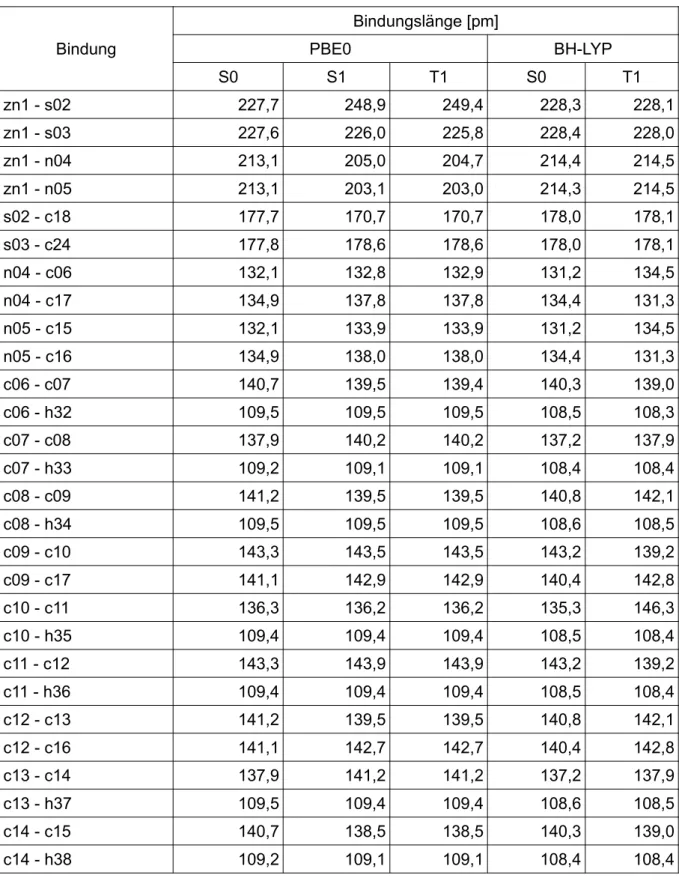
Bei den Konformeren, welche mit dem gleichen Funktional optimiert wurden, sind die Abstände innerhalb der Kohlenstoffringe relativ ähnlich. Die meisten Bindungslängen unterscheiden sich um maximal 0,1 pm. Der größte Unterschied bei den Bindungs- längen innerhalb der Kohlenstoffringe beträgt 0,4 pm. Dahingegen unterscheiden sich die Bindungslängen zwischen Schwefel und Zink, beziehungsweise zwischen Stickstoff und Zink, meist relativ stark mit Unterschieden bis zu 3,8 pm. Werden die Konformere verglichen, welche mit unterschiedlichen Funktionalen optimiert wurden, fällt auf, dass die Bindungslängen sich stärker unterscheiden als bei dem Vergleich innerhalb der Funktionale. Die größte Differenz beträgt dabei 1,5 pm.
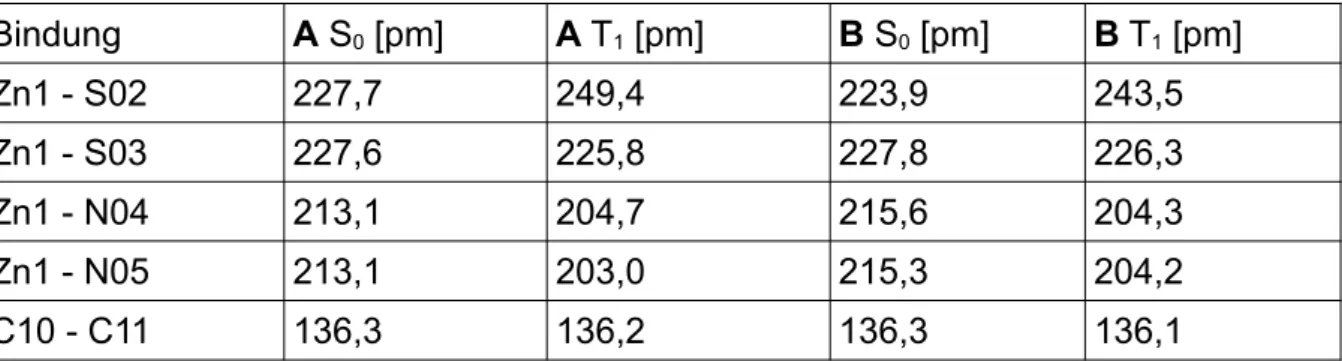
Bei dem Vergleich zu den Literaturdaten [41] beträgt die größte Abweichung zu dem Konformer A, welches mit PBE0 optimiert wurde, 3,0 pm. Bei dem mit BH-LYP opti- mierten, beträgt die größte Abweichung 4,3 pm. Die größte Abweichung zwischen dem Konformer B, welches mit PBE0 optimiert wurde, und der Literatur beträgt 5,5 pm. Bei dem mit BH-LYP optimierten beträgt die größte Abweichung 7,0 pm. Bei bei- den Konformeren und beiden Funktionalen liegt diese Abweichung bei der Bindung Zn1 - N04 vor. (Tab. 1)
Tab. 1: Vergleich der Bindungslängen der beiden Konformere und der Literatur [41] im Grundzustand
Bindung PBE0 BH-LYP
Literatur [pm]
A [pm] B [pm] A [pm] B [pm]
Zn1 - S02 227,7 223,9 228,3 224,7 225,9
Zn1 - S03 227,6 227,8 228,4 228,1 227,3
Zn1 - N04 213,1 215,6 214,4 217,1 210,1
Zn1 - N05 213,1 215,3 214,3 216,7 211,7
Bei dem Vergleich der Konformere, welche mit dem selben Funktional optimiert wur-
den, ist zu erkennen, dass die Bindungswinkel innerhalb der Kohlenstoffringe maxi-
mal 0,8° von einander abweichen, auch die, die über die Stickstoffe aufgespannt wer-
den. Dahingegen weichen die Bindungswinkel, welche vom Zink ausgehen oder über
das Zink hinweg gehen, stärker voneinander ab um bis zu 15°. Werden die Konfor-
mere, welche mit unterschiedlichen Funktionalen optimiert wurden, verglichen, ist
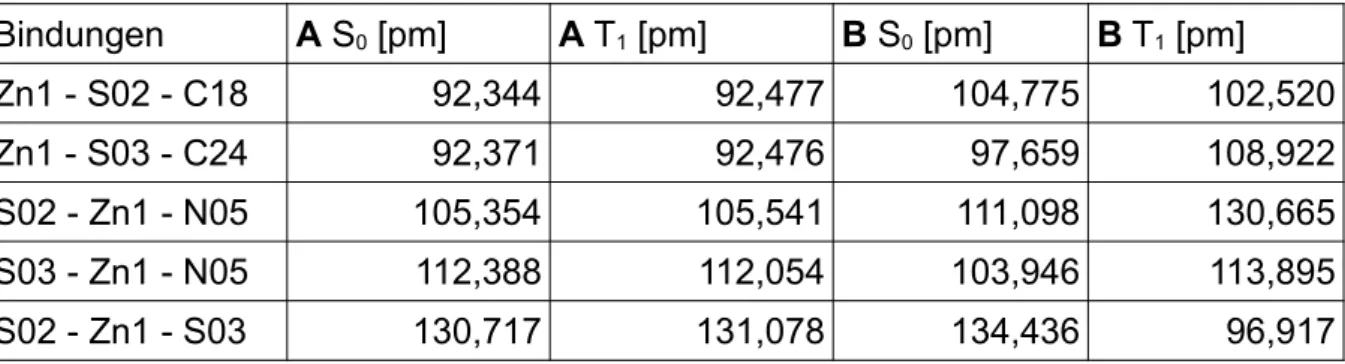
auffällig, dass bei Konformer A nur zwei Bindungswinkel um mehr als 0,8° abwei- chen. Die jeweiligen Abweichungen der beiden Winkel von Zink über N05 zu einem der danebenliegenden Kohlenstoffen beträgt ca. 15,1°. Bei dem Vergleich der Kon- formere B, welche mit unterschiedlichen Funktionalen optimiert wurden, weicht kein Bindungswinkel um mehr als 1,0° ab.
Bei dem Vergleich mit der Literatur mit dem Konformer A, welches mit PBE0 opti- miert wurde, beträgt die größte Abweichung 12,94°. Die größten Abweichungen sind bei Winkeln, die sowohl den Schwefel S03, als auch Zink enthalten. Diese Abwei- chungen treten auch bei Konformer B, welches mit PBE0 optimiert wurde, auf, dort beträgt die größte Abweichung allerdings nur 8,65°. Bei den Konformeren, welche mit BH-LYP optimiert wurden, beträgt die größte Abweichung bei Konformer A 16,24°
und liegt bei Zink und Stickstoff N05 vor. Bei Konformer B liegen ähnliche Abwei- chungen, wie bei dem PBE0-Funktional vor. Dort beträgt die größte Abweichung mit 8,59° der Bindungswinkel vom Schwefel zum Zink zum anderen Schwefel vor.
Die wichtigsten Bindungswinkel im Vergleich sind in Tabelle 2 zu sehen.
Tab. 2: Vergleich der Bindungswinkel der beiden Konformere und der Literatur [41] im Grund- zustand
Bindungen PBE0 BH-LYP
Literatur [°]
A [°] B [°] A [°] B [°]
Zn1 - S02 - C18 91,586 104,875 92,344 104,775 104,52
Zn1 - S03 - C24 91,600 97,878 92,371 97,659 95,00
S02 - Zn1 - N05 105,044 112,037 105,354 111,098 114,50 S03 - Zn1 - N05 112,102 103,495 112,388 103,946 106,90 Bei den Diederwinkeln zeigt sich ein ähnlicher Trend. Die Diederwinkel unterschei- den sich innerhalb der Kohlenstoffringe kaum. Die Differenz der Diederwinkel ist je- doch groß, wenn diese über das Zinkatom aufgespannt werden. Wenn allerdings die Konformere der unterschiedlichen Funktionalen gegenüber gestellt werden, sind die Diederwinkel recht ähnlich und weichen kaum voneinander ab.
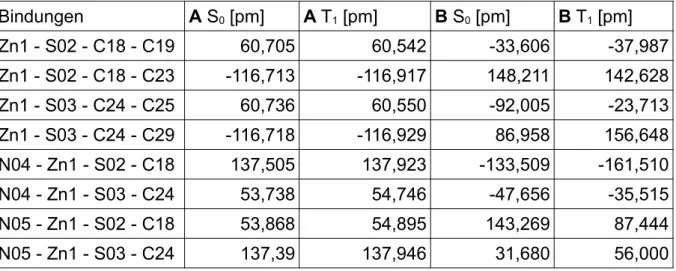
Die wichtigsten Diederwinkel sind in Tabelle 3 zu sehen.
Tab. 3: Vergleich der Diederwinkel der beiden Konformere im Grundzustand
Bindungen PBE0 BH-LYP
A [°] B [°] A [°] B [°]
Zn1 - S02 - C18 - C19 59,977 -30,248 60,705 -33,606
Zn1 - S02 - C18 - C23 -117,653 151,650 -116,713 148,211
Zn1 - S03 - C24 - C25 60,029 -92,626 60,736 -92,005
Zn1 - S03 - C24 - C29 -117,643 87,070 -116,718 86,958 N04 - Zn1 - S02 - C18 138,417 -134,014 137,505 -133,509
N04 - Zn1 - S03 - C24 54,184 -50,235 53,738 -47,656
N05 - Zn1 - S02 - C18 54,430 141,867 53,868 143,269
N05 - Zn1 - S03 - C24 138,337 29,525 137,39 31,680
6.2. Vergleich der Konformere im ersten angeregten Triplettzustand (LLCT)
Für beide Konformere wurde der LLCT T
1-Zustand mittels PBE0 optimiert. Abbildung 6 zeigt die grafische Darstellung der Konformere A (links) und B (rechts).
Für die Optimierung mittels BH-LYP zeigte zuerst eine lokale Anregung des 1,10-
Phenanthrolin im T
1-Zustand für beide Konformere in der ESCF-Rechnung. Bei der
Geometrieoptimierung dieses Zustands zeigt Konformer A im T
1-Zustand noch eine
lokale Anregung, wohingegen Konformer B erst im T
2-Zustand eine lokale Anregung
zeigt. Nach der DFT/MRCI-Rechnung weist das Konformer A erst im T
3-Zustand eine
lokale Anregung auf. Dahingegen zeigt das Konformer B bis zum T
10-Zustand keine
lokale Anregung. Die Optimierung der lokalen Anregung ist bei beiden Konformeren
nicht gelungen.
Abb. 6: Darstellung der optimierten LLCT T1 Geometrie mit PBE0 zweier Konformere des Zn(II)(4-CH3-C6H4S)2(1,10-Phenanthrolin), links Konformer A, rechts Konformer B
Bei dem Vergleich der beiden LLCT T
1Zustände, der mit PBE0 optimierten Konfor- meren, fällt auf, dass die Bindungslängen meist ähnlich sind. Die größte Differenz zwischen den Abständen besteht bei dem Schwefel, welcher sich bei Konformer A nach hinten oben raus gedreht hat, und dem oberen Schwefel des Konformers B.
Der Unterschied beträgt dort 5,9 pm. Es weichen nur 6 Bindungslängen im Vergleich zwischen den beiden Konformeren im LLCT T
1-Zustand um 0,5 pm oder mehr als das ab. Bei den Konformeren, welche mit dem Funktional BH-LYP optimiert wurden, sind die Bindungslängen nicht so ähnlich. Dort weicht ca. die Hälfte der Bindungslän- gen um mehr als 0,5 pm ab.
Werden die Konformere im LLCT T
1-Zustand und dem Grundzustand verglichen, ist
auffällig, dass bei der Optimierung mit PBE0 das Konformer A seine Struktur stark
verändert. Einer der beiden Thiophenolringe dreht sich von dem 1,10-Phenanthrolin
weg. Der Thiophenolring steht dann orthogonal zum 1,10-Phenanthrolinring. Dadurch
nähert sich diese Struktur der des Konformers B an. Aufgrund dessen verändern sich
die Bindungslängen vor allem um das Zinkatom herum zwischen den beiden Zustän-
den. Die größte Differenz ist bei der Bindungslänge zwischen dem Schwefel, welcher
Schwefel, welcher von dem 1,10-Phenanthrolin weggedreht ist. Die Differenz beträgt 19,6 pm.
Bei der Optimierung mit BH-LYP verändert allerdings Konformer B seine Struktur und nähert sich der Struktur des Konformers A an. Dabei dreht sich der Thiophenolring, welcher sich unter das 1,10-Phenanthrolin gedreht hat, etwas raus. Dort verändern sich die Bindungslängen um bis zu 18,5 pm. Bei dem Konformer A ist die größte Än- derung der Bindungslänge in dem 1,10-Phenanthrolinring mit 11 pm. Die wichtigsten Bindungslängen und diese mit der größten Abweichung sind in Tabellen 5 und 6 zu sehen.
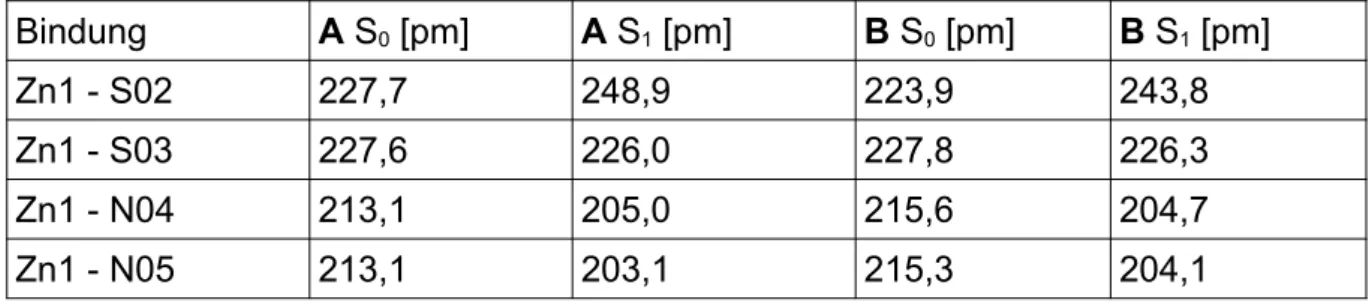
Tab. 5: Vergleich der Bindungslängen der Konformere im Grundzustand und ersten angereg- tem Triplett-Zustand mit dem Funktional PBE0
Bindung
A S0[pm]
A T1[pm]
B S0[pm]
B T1[pm]
Zn1 - S02 227,7 249,4 223,9 243,5
Zn1 - S03 227,6 225,8 227,8 226,3
Zn1 - N04 213,1 204,7 215,6 204,3
Zn1 - N05 213,1 203,0 215,3 204,2
C10 - C11 136,3 136,2 136,3 136,1
Tab. 6: Vergleich der Bindungslängen der Konformere im Grundzustand und ersten angereg- tem Triplett-Zustand mit dem Funktional BH-LYP
Bindung
A S0[pm]
A T1[pm]
B S0[pm]
B T1[pm]
Zn1 - S02 228,3 228,1 224,7 227,1
Zn1 - S03 228,4 228,0 228,1 246,6
Zn1 - N04 214,4 214,5 217,1 202,4
Zn1 - N05 214,3 214,5 216,7 203,3
C10 - C11 135,3 146,3 135,3 135,1
Die Bindungswinkel in dem T
1-Zustand, welche mit PBE0 optimiert wurden, sind rela-
tiv ähnlich, die maximale Abweichung beträgt 0,8°. Nur bei den Winkeln, welche Zink
und mindestens eines der beiden Schwefelatome beinhalten, weichen um bis zu
10,6° voneinander ab. Bei den Konformeren, welche mit dem BH-LYP optimiert wur-
den, weichen die Bindungswinkel stärker voneinander ab. Die größte Abweichung
besteht bei dem Winkel von Schwefel über Zink zu dem anderen Schwefel mit ca.
34,2°.
Werden die LLCT T
1-Strukturen mit dem Grundzustand verglichen, sind bei den mit PBE0 optimierten Konformeren die Abweichungen der Bindungswinkel am größten, sobald das Zink einbezogen ist. Hierbei beträgt die größte Abweichung fast 24°. Die Winkel innerhalb der Kohlenstoffringe sind relativ ähnlich geblieben. Dieser Trend ist auch bei dem mit BH-LYP optimierten Konformer B gegeben. Dort beträgt die größte Abweichung allerdings fast 37,6°. Bei dem mit BH-LYP optimierten Konformer A wei- chen nur zwei Bindungswinkel stark voneinander ab. Diese sind von Zink über Stick- stoff N05 und zu einem der beiden Kohlenstoffe daneben aufgespannt. Diese wei- chen um bis zu 14,2° voneinander ab. (Tab. 7,8)
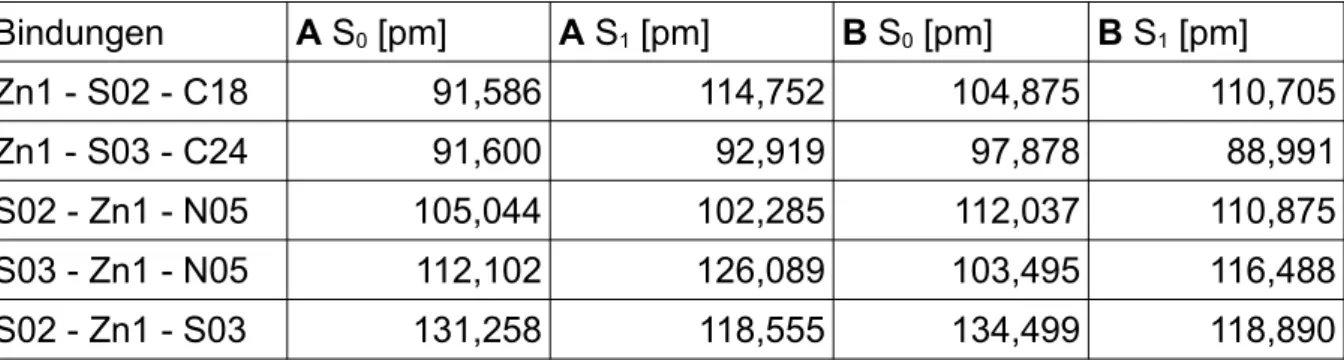
Tab. 7: Vergleich der Bindungswinkel der Konformere im Grundzustand und ersten angereg- tem Triplett-Zustand mit dem Funktional PBE0
Bindungen
A S0[pm]
A T1[pm]
B S0[pm]
B T1[pm]
Zn1 - S02 - C18 91,586 114,808 104,875 110,354
Zn1 - S03 - C24 91,600 92,908 97,878 88,936
S02 - Zn1 - N05 105,044 101,779 112,037 110,862
S03 - Zn1 - N05 112,102 126,511 103,495 115,921
S02 - Zn1 - S03 131,258 118,371 134,499 118,544
Tab. 8: Vergleich der Bindungswinkel der Konformere im Grundzustand und ersten angereg- tem Triplett-Zustand mit dem Funktional BH-LYP
Bindungen
A S0[pm]
A T1[pm]
B S0[pm]
B T1[pm]
Zn1 - S02 - C18 92,344 92,477 104,775 102,520
Zn1 - S03 - C24 92,371 92,476 97,659 108,922
S02 - Zn1 - N05 105,354 105,541 111,098 130,665
S03 - Zn1 - N05 112,388 112,054 103,946 113,895
S02 - Zn1 - S03 130,717 131,078 134,436 96,917
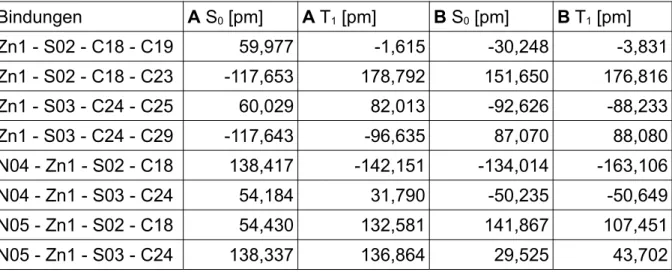
Bei den Diederwinkeln unterscheiden sich die Winkel innerhalb der Kohlenstoffringe relativ wenig. Dahingegen unterscheiden sie sich in dem Bereich um das Zinkatom, bei den mit PBE0 optimierten Konformeren, stark mit bis zu 175,3°. Bei den mit BH- LYP optimierten Konformeren unterscheiden sich die Diederwinkel mit dem Zinkatom um maximal 80,0°.
Bei dem Vergleich des Konformers A von dem ersten angeregten Triplett-Zustand mit dem Grundzustand, welche beide mit PBE0 optimiert wurden, ändern sich am stärks- ten mit fast 79,5° von Stickstoff über Zink zu Schwefel, welcher sich von dem 1,10- Phenanthrolin wegdreht, und dem Kohlenstoff. Bei dem Vergleich des Konformers A, welches mit BH-LYP optimiert wurde, fällt auf, dass die Diederwinkel sich um nicht mehr als 1,1° unterscheiden.
Bei dem Vergleich des Konformers B, welches mit PBE0 optimiert wurde, weicht der gleiche Diederwinkels am stärksten von der Grundzustandsgeometrie, wie bei dem Konformer A, ab. Bei dem mit BH-LYP optimiert Konformer, sind die Diederwinkel am unterschiedlichsten, welche über Zink, dem Schwefel, welcher unterhalb des 1,10- Phenanthrolin liegt, und den folgenden Kohlenstoffatomen aufgespannt sind. Dort be- trägt die Differenz ca. 69,7°. Das Verhalten der Diederwinkel wird in den Tabellen 9 und 10 aufgezeigt.
Tab. 9: Vergleich der Bindungswinkel der Konformere im Grundzustand und ersten angereg- tem Triplett-Zustand mit dem Funktional PBE0
Bindungen
A S0[pm]
A T1[pm]
B S0[pm]
B T1[pm]
Zn1 - S02 - C18 - C19 59,977 -1,615 -30,248 -3,831
Zn1 - S02 - C18 - C23 -117,653 178,792 151,650 176,816
Zn1 - S03 - C24 - C25 60,029 82,013 -92,626 -88,233
Zn1 - S03 - C24 - C29 -117,643 -96,635 87,070 88,080 N04 - Zn1 - S02 - C18 138,417 -142,151 -134,014 -163,106
N04 - Zn1 - S03 - C24 54,184 31,790 -50,235 -50,649
N05 - Zn1 - S02 - C18 54,430 132,581 141,867 107,451
N05 - Zn1 - S03 - C24 138,337 136,864 29,525 43,702
Tab. 10: Vergleich der Bindungswinkel der Konformere im Grundzustand und ersten ange- regtem Triplett-Zustand mit dem Funktional BH-LYP
Bindungen
A S0[pm]
A T1[pm]
B S0[pm]
B T1[pm]
Zn1 - S02 - C18 - C19 60,705 60,542 -33,606 -37,987
Zn1 - S02 - C18 - C23 -116,713 -116,917 148,211 142,628
Zn1 - S03 - C24 - C25 60,736 60,550 -92,005 -23,713
Zn1 - S03 - C24 - C29 -116,718 -116,929 86,958 156,648 N04 - Zn1 - S02 - C18 137,505 137,923 -133,509 -161,510
N04 - Zn1 - S03 - C24 53,738 54,746 -47,656 -35,515
N05 - Zn1 - S02 - C18 53,868 54,895 143,269 87,444
N05 - Zn1 - S03 - C24 137,39 137,946 31,680 56,000
6.3. Vergleich der Konformere im ersten angeregten Singulettzustand
Für beide Konformere wurde ebenso der erste angeregte Singulett-Zustand berech- net. Dieser ist ähnlich in Geometrie und Orbitalverhalten zu dem T
1-Zustand (LLCT).
Abb. 8: Darstellung der optimierten S1 Geometrie mit PBE0 zweier Konformere des Zn(II)(4-CH3-C6H4S)2(1,10-Phenanthrolin), links Konformer A, rechts Konformer B
Bei dem Vergleich der S
1-Geometrien ist auffällig, dass nur die Bindungslänge vom Zink zu dem Schwefelatom, welches sich von dem 1,10-Phenanthrolin weg dreht, stark mit 5,1 pm unterscheidet. Dieses ist ähnlich zu dem Vergleich von den T
1-Geo- metrien. Die zweitgrößte Abweichung ist zwischen dem Zink und dem Stickstoffatom, N05 mit 1,0 pm.
Auch die Unterschiede zum Grundzustand sind bei beiden Konformeren ähnlich zu dem Vergleich zwischen T
1- und Grundzustand. Die größte Differenz zwischen den Zuständen ist bei dem Konformer A zwischen Schwefel S02 und Zink mit 21,2 pm.
Die nächstgrößere Abweichung besteht bei Zink und den Stickstoffen mit bis zu 10,0 pm. Bei dem Konformer B sind diese ebenfalls die größten Abweichungen. Allerdings sind die Differenzen zwischen Zink und den Stickstoffen bis zu 11,2 pm groß. (Tab.
11)
Tab. 11: Vergleich der Bindungslängen der Konformere im Grundzustand und ersten ange- regtem Singulett-Zustand mit dem Funktional PBE0
Bindung
A S0[pm]
A S1[pm]
B S0[pm]
B S1[pm]
Zn1 - S02 227,7 248,9 223,9 243,8
Zn1 - S03 227,6 226,0 227,8 226,3
Zn1 - N04 213,1 205,0 215,6 204,7
Zn1 - N05 213,1 203,1 215,3 204,1
Die Bindungswinkel zwischen den Konformeren im S
1-Zustand unterscheiden sich le- diglich, wenn der Winkel über mindestens einem Schwefel und dem Zinkatom aufge- spannt wird. Dabei beträgt die maximale Abweichung 9,6°. Die anderen Winkel wei- chen zwischen den Konformeren um nicht einmal 1,0° ab.
Bei dem Vergleich zwischen dem Konformer A in Grundzustandsgeometrie und S
1-
Geometrie liegt die größte Differenz zwischen Zink, Schwefel S02 und dem folgen-
den Kohlenstoff bei ca. 23,2°. Die nächstgrößeren Abweichungen mit fast 14,0° tre-
ten auf, wenn der Winkel Schwefel, Zink und Stickstoff beinhaltet. Bei dem Konfor-
mer B ist die größte Winkeländerung zwischen Schwefel, Zink und dem anderen
Schwefel. Diese beträgt ca. 15,7°. Verdeutlicht werden die wichtigsten Bindungswin-
kel in Tabelle 12.
Tab. 12: Vergleich der Bindungswinkel der Konformere im Grundzustand und ersten ange- regtem Singulett-Zustand mit dem Funktional PBE0
Bindungen
A S0[pm]
A S1[pm]
B S0[pm]
B S1[pm]
Zn1 - S02 - C18 91,586 114,752 104,875 110,705
Zn1 - S03 - C24 91,600 92,919 97,878 88,991
S02 - Zn1 - N05 105,044 102,285 112,037 110,875
S03 - Zn1 - N05 112,102 126,089 103,495 116,488
S02 - Zn1 - S03 131,258 118,555 134,499 118,890
Aufgrund der unterschiedlichen räumlichen Struktur des S
1-Zustandes der Konforme- re, weichen die Diederwinkel, welche das Zinkatom enthalten, meist stark voneinan- der ab. Die maximale Abweichung beträgt 174,5°. Es gibt insgesamt 5 Diederwinkel, die mehr als 80° abweichen. Die anderen Diederwinkel sind relativ ähnlich und wei- chen maximal um 18,5° ab.
Wird die erste angeregte Singulett-Geometrie des Konformers A mit dem Grundzu- stand verglichen, ist die größte und zweitgrößte Abweichung mit bis zu 80,2° zwi- schen einem Stickstoff, Zink, Schwefel, welcher sich von dem 1,10-Phenanthrolin wegdreht, und dem folgenden Kohlenstoff. Bei dem Konformer B beträgt die größte Abweichung 27,6° und wird über die beiden Schwefelatome aufgespannt. (Tab. 13)
Tab. 13: Vergleich der Bindungswinkel der Konformere im Grundzustand und ersten ange- regtem Singulett-Zustand mit dem Funktional PBE0Bindungen
A S0[pm]
A S1[pm]
B S0[pm]
B S1[pm]
Zn1 - S02 - C18 - C19 59,977 -2,323 -30,248 -7,281
Zn1 - S02 - C18 - C23 -117,653 178,155 151,650 173,625
Zn1 - S03 - C24 - C25 60,029 82,045 -92,626 -87,361
Zn1 - S03 - C24 - C29 -117,643 -96,573 87,070 88,963 N04 - Zn1 - S02 - C18 138,417 -141,420 -134,014 -156,023
N04 - Zn1 - S03 - C24 54,184 32,052 -50,235 -50,517
N05 - Zn1 - S02 - C18 54,430 133,232 141,867 114,778
N05 - Zn1 - S03 - C24 138,337 136,227 29,525 43,719
6.4. Anregungen und Emissionen
Für die Konformere sind DFT / MRCI-Rechnungen in Grundzustands- und angereg- ten Zustandsgeometrien durchgeführt worden. Dabei wurden die Arten der Anregun- gen und deren benötigten Energien berechnet. Bei dem Vergleich der Anregungs- energien ist auffällig, dass der Unterschied zwischen den Konformeren im T
1größer ist als im S
0.
In Tabelle 14 werden die Anregungsenergien der ersten neun angeregten Singulett- und ersten zehn Triplettzustände der beiden Konformeren an der S
0Geometrie dar- gestellt.
Tab. 14: Vergleich der Anregungsenergien und Oszillatorstärken von der mit Avogadro er- stellten Struktur und von der optimierten Literaturstruktur an der Grundzustandsgeometrie
Anregung ΔE [eV] A Oszillatorstärke A ΔE [eV] B Oszillatorstärke B S0
S0 → S1 2,4641 0,01159 2,3186 0,00204
S0 → S2 2,5506 0,02028 2,4624 0,00110
S0 → S3 2,6587 0,00254 2,8107 0,02480
S0 → S4 2,7517 0,00153 2,9374 0,01119
S0 → S5 3,0111 0,02211 2,9483 0,00341
S0 → S6 3,1873 0,00152 3,0944 0,00079
S0 → S7 3,7329 0,00135 3,6869 0,00041
S0 → S8 3,8918 0,00785 3,8372 0,01068
S0 → S9 3,9075 0,00028 3,9953 0,00251
S0 → T1 2,3924 2,2719
S0 → T2 2,4891 2,4220
S0 → T3 2,6161 2,7532
S0 → T4 2,7023 2,8809
S0 → T5 2,9459 2,9015
S0 → T6 3,1310 3,0587
S0 → T7 3,3462 3,3495
S0 → T8 3,6599 3,6517
S0 → T9 3,6813 3,6534
S0 → T10 3,8070 3,7993
Die beiden Konformere haben verschiedene Oszillatorstärken. Bei dem Vergleich wird deutlich, dass die Anregungen unterschiedlich hell sind. Bei Konformer A ist die hellste Anregung jene vom S
0in den S
25, bei Konformer B ist es der Übergang von S
0in den S
23Zustand. Diese Übergänge sind in Abbildung 9 dargestellt. Die Übergänge sind für das Maximum des Absorptionsspektrum (Abb. 10) verantwortlich. In Konfor- mer A ist eine LLCT Anregung für das Maximum in dem Absorptionsspektrum verant- wortlich, in Konformer B eine lokale Anregung auf dem 1,10-Phenanthrolin gemischt mit einer LLCT Anregung.
Abb. 9: Darstellung der Differenzdichten der Zustände, die für die hellen Übergänge in dem Absorptionsspektrum zu sehen sind, links Konformer A S25 → S0, rechts Konformer B
S23 → S0
Mithilfe der Oszillatorstärken kann ein Absorptionsspektrum berechnet werden. In
den folgenden Spektren sind die Absorptionsspektren von dem mit Avogadro erstell-
ten Konformer, der optimierten Literaturstruktur, sowie einem Spektrum aus der Lite-
ratur dargestellt (Abb. 10, 11).
Abb. 10: Darstellung der Absorptionsspektren [43] des Konformers A (Optimierung mit PBE0) aufgenommen im Vakuum (türkis), in Ethanol (lila), in Chlormethan (grün) verglichen
mit dem digitalisierten Spektrum aus der Literatur [10]
Abb. 11: Darstellung der Absorptionsspektren [43] des Konformers B (Optimierung mit PBE0) aufgenommen im Vakuum (türkis), in Ethanol (lila), in Chlormethan (grün) verglichen mit dem
digitalisierten Spektrum aus der Literatur [10]
Da in der Literatur kein Absorptionsspektrum für (4-CH
3-C
6H
4S)
2Zn(1,10-Phenanthro-
lin) vorhanden ist, wurde für das Absorptionsspektrum aus der Literatur [10] ein
Spektrum von Zn(4-ClC
6H
4S)
2(1,10-Phenanthrolin) genommen. Allerdings wurde von
Highland et. al. 1986 [9] festgestellt, dass die beiden Komplexe (4-Methyl-Thiophenol
und 4-Chlor-Thiophenol) sich bei den Absorptionen und Emissionen kaum unter- scheiden [9].
Das Spektrum aus der Literatur wurde in einem Lösemittelgemisch aus Chlormethan und Ethanol (1:19, v/v) aufgenommen.
Die berechneten Absorptionsspektren basieren auf Rechnungen der Anregungsener- gien der DFT/MRCI Rechnung mit jeweils 30 Wurzeln und wurden auf deren Grund- lage erstellt. Die Gaußverbreiterung beträgt für die berechneten Absorptionssprek- tren 1500 cm
-1. [43]
Im Vergleich zwischen den berechneten und dem Literaturabsorptionsspektrum fällt auf, dass die berechnete Absorptionsspektren nicht so stark im Bereich des Maxi- mums voneinander abweichen. Bei den Berechnungen im Vakuum weichen die Maxi- ma bei beiden Konformeren am stärksten ab. Bei Konformer A ist das berechnete Absorptionsspektrum in Chlormethan nah zu dem experimentell bestimmten Spek- trum. Bei Konformer B ähneln die berechneten Absorptionsspektren im Maximum weniger dem experimentellen Absorptionsspektrum. Die berechneten Absorptions- spektren haben ihr Maximum bei 256,6 nm (Konformer B) beziehungsweise bei 257,1 nm (Konformer A). Dahingegen liegt das Maximum der Literatur [10] bei etwa 267 nm. Für das Konformer A ist die Anregung in den S
25und bei dem Konformer B in den S
23entscheidend für die Maxima. In Abbildung 9 sind die Differenzdichten der optimierten Konformere A und B mittels PBE0 zu sehen.
Ebenso fällt das berechnete Absorptionsspektrum des Konformers B schneller ab als das Absorptionsspektrum aus der Literatur [10]. Das Absorptionsspektrum des Kon- formers A nähert sich dem Spektrum der Literatur an.
Für die Optimierung mittels BH-LYP sind die Absorptionsspektren ähnlich. Die Ab- sorptionsspektren sind im Anhang in Abbildung B13 zu sehen.
Die Wahl der Lösemittelumgebung hat einen starken Einfluss auf die Lage der ange- regten Zustände (Abb. 10, 11). Die lokalen Anregungen auf dem 1,10-Phenanthrolin im kurzwelligeren Bereich werden im Vakuum bessere beschrieben als die langwelli- geren LLCT Anregungen. Dieses spiegelt die Ergebnisse aus der Literatur wider. [44]
Für die berechneten Lösemittel [43] werden die langwelligeren LLCT Zustände ener-
Wird der erste angeregte Zustand mit dem Grundzustand des jeweiligen Konformers verglichen, fällt auf, dass das Dipolmoment bei dem Konformer A seine Richtung um- kehrt und sich der Betrag nahe zu halbiert. Bei dem Konformer B kehrt das Dipolmo- ment seine Richtung fast komplett um, wohingegen der Betrag gleich bleibt. Das hat dahingehend eine Bedeutung für das Lösemittel, da das Solvens sich an dem Dipol- moment vom Grundzustand des Solvats orientiert. Somit stabilisiert das Lösemittel den Komplex in Lösung. Ändert sich das Dipolmoment bei der Anregung, wie bei dem vorliegendem Komplex bei den CT Zuständen, kann das Lösemittel den Kom- plex in der Lösung nicht mehr stabilisieren. Aufgrund dessen, dass die Absorptions- prozesse ca. 10
-15s benötigen, aber die Relaxationszeit für die Dipolorientierung im Lösemittel bei ca. 10
-11s liegt. Deshalb ist bei der Absorption die Lösemittelumge- bung dieselbe, die auch bei dem elektronischen Grundzustand vorliegt. [45]
Bei der Emission von langlebigeren angeregten Zuständen ist die Dipolorientierung des Lösemittels relaxiert. Das bedeutet für den Komplex, dass dieser sich im Grund- zustand befindet, aber die Lösemittelumgebung noch an den angeregten Zustand angepasst ist. Dadurch kommt es auch hierbei zu einer Verschiebung der CT Zustän- de in den kurzwelligeren Bereich. [44,45]
In dem Energiediagramm, welches in Abbildung 12 dargestellt ist, sind die Zustände
im Vakuum aufgetragen, welche die niedrigliegensten CT und lokalen Anregungen
zeigen. Darin wird deutlich, dass die Konformere optimiert mit PBE0 sich auch ener-
getisch, sowohl im Grundzustand wie auch im ersten angeregten Triplett-Zustand un-
terscheiden.
Abb. 12: Darstellung der energetischen Lage der Übergänge an den PBE0 optimierten Kon- formeren im Vakuum, die Buchstaben P und T deuten an, dass dort die Orbitale lokalisiert
sind, P steht für 1,10-Phenanthrolin, T für Thiophenol.
In der Abbildung 12 wird dargestellt in welcher Relation die Zustände innerhalb der Konformere stehen. In beiden Konformeren werden an der T
1-Geometrie im Ver- gleich zur S
0-Geometrie die Charge-Transfer Übergänge energetisch abgesenkt. Die meisten lokalen Anregungen werden auch abgesenkt, allerdings nicht so stark wie die Charge-Transfer Zustände.
Einige lokalen Anregungen werden im Gegensatz zu den anderen Zuständen gering angehoben. Vor allem die lokale Triplett-Anregung auf dem Thiophenol wird in der mit Avogadro erstellten Struktur um etwa 1,23 eV angehoben und liegt damit über der Singulett-Anregung, welche auf dem Thiophenol lokalisiert ist.
Die Lage der Charge-Transfer Zustände ist in beiden Konformeren relativ ähnlich.
Bei den lokalen Anregungen gibt es größere Unterschiede, sowohl in der energeti- schen Lage als auch in der Reihenfolge der Zustände.
0 1 2 3 4 5
Energie [eV]
1pp* (T)
1pp* (P)
3pp* (P)
3pp* (P)
3pp* (P)
1pp* (T)
3pp* (P)
1pp* (P)
3pp* (T) 1pp* (P)
1pp* (P) 1pp* (T)
1pp* (T)
3pp* (T)
3pp* (T)
1
CT
1
CT
1
CT
3
CT
1
CT
3
CT
3CT
3
CT
T1 S0
T1 S0
Konformer B
Konformer A
Bei Konformer A ist die Reihenfolge der Zustände in der T
1-Geometrie, bis auf den
3pp* Zustand am Thiophenol, in der selben Reihenfolge wie die Zustände an der S0
- Geometrie.
Am Grundzustand des Konformers B weisen die berechneten Triplett-Zustände kei- nen lokalen Anregungscharakter am Thiophenol auf. An der T
1-Geometrie liegt der Zustand mit lokalem Anregungscharakter am Thiophenol oberhalb des
3pp* Zustan-des, der auf dem 1,10-Phenanthrolin lokalisiert ist. Dadurch ist der Abstand an der T
1-Geometrie zwischen dem Singulett- und dem Triplett-Zustand, der auf dem Thio- phenol lokalisierten Anregungen, geringer als bei den Zuständen, der auf dem 1,10- Phenanthrolin lokalisierten Anregungen. Der Abstand zwischen den lokalen Anregun- gen auf dem Thiophenol liegt bei 0,73 eV, zwischen den Anregungen auf dem 1,10- Phenanthrolin bei 1,01 eV.
Bei dem mit BH-LYP optimierten Konformeren wurden die CT Zustände gegenüber den lokalen Anregungen energetisch weiter abgesenkt. Das Energiediagramm ist in Abbildung B15 im Anhang zu erkennen.
Mithilfe des VIBES-Programms wurden die Emissionsspektren von dem T
1-Zustand in den Grundzustand berechnet. Bei einer Temperatur von 298 K konnte bei beiden Konformeren keine Emission berechnet werden, wohingegen bei 77 K am Konformer
B, welches mit PBE0 optimiert wurde, eine Emission zu erkennen ist. Die Emissiondes Komplexes ist temperaturabhängig und ist nicht bei hohen Temperaturen zu er- kennen. Dieses Emissionsspektrum wird in Abbildung 13 dargestellt. Es ist kein Emissionsspektrum von Konformer A dargestellt, welches mit PBE0 optimiert wurde, da diese eine starke Verdrillung in der T
1- im Gegensatz zu der S
0-Geometrie auf- weist. Aus diesem Grund kann kein Emissionsspektrum berechnet werden.
Bei den mit BH-LYP optimierten Konformeren konnte nur bei dem Konformer A ein
Emissionsspektrum erstellt werden. Dieses ähnelt in keiner Weise dem der Literatur
[8,10,11]. Das Emissionsspektrum ist in Abbildung B14 im Anhang zu sehen.
Abb. 13: Darstellung des Emissionsspektrums bei 77 K von Konformer B optimiert mit PBE0
![Tab. 2: Vergleich der Bindungswinkel der beiden Konformere und der Literatur [41] im Grund- Grund-zustand](https://thumb-eu.123doks.com/thumbv2/1library_info/4529778.1596139/25.892.112.788.679.858/tab-vergleich-bindungswinkel-konformere-literatur-grund-grund-zustand.webp)