zum Thema
Temperaturabhängige Untersuchungen von Uran-Redoxreaktionen in halidhaltigen Lösungen
Technische Universität Dresden
Fakultät Mathematik und Naturwissenschaften
Fachrichtung Chemie und Lebensmittelchemie
Professur für Radiochemie / Radioökologie
Eingereicht von Maximilian Demnitz Geb. am 16.03.1994 in Dresden
Gutachter: Prof. Dr. Thorsten Stumpf Betreuer: Dr. Nina Maria Huittinen
Dresden, den 17.09.2018
Angefertigt am:
Institut für Ressourcenökologie Helmholtz-Zentrum Dresden-Rossendorf
Die vorliegende Arbeit wurde am Helmholtz-Zentrum Dresden-Rossendorf am Institut für Ressourcenökologie angefertigt. Mein Dank gilt Herrn Professor Doktor Thorsten Stumpf für die Möglichkeit, die vorliegende Arbeit am Institut angefertigt haben zu dürfen.
Frau Dr. Nina Maria Huittinen möchte ich für die exzellente und freundliche Betreuung mit allen damit verbundenen Arbeiten während des zuvor durchgeführten Forschungspraktikums und während der Masterarbeit danken.
Für die kontinuierliche Unterstützung bei dem Aufbau des Lasersystems zur Detektion von Uran(VI) und der Methodenentwicklung möchte ich Herrn Henry Lösch danken, welcher sich immer Zeit genommen hat, um auftretende Fragen zu klären und bei den Aufbauarbeiten behilflich zu sein.
Besonderer Dank gilt Herrn Dirk Falkenberg, welcher die konzeptionellen Darstellungen der entwickelten Messzelle verfeinerte und diese nach Wunschvorstellungen fertigte.
Herrn Dr. Robin Steudtner möchte ich danken für die Unterstützung bei allen Fragen, welche in Bezug auf Laser, Uran oder technische Aspekte angefallen sind.
Vielen Dank gebührt ebenso Herrn Thomas Zimmermann, welcher mich bei der Bedienung des Programms Medusa unterstützte, das Uran(IV)-Lasersystem vorstellte und exzellente Uran(IV)-UV/VIS-Referenzspektren zu Verfügung stellte.
Weiterhin möchte ich Frau Doktor Astrid Barkleit danken, welche eine tiefere Einführung in das Uran(IV)-Lasersystem ermöglichte und Einstellungen vornahm, welche das Messen ermöglichten.
Großer Dank gilt allen Mitarbeiterinnen und Mitarbeitern am Institut für Ressourcenökologie und am HZDR, welche mir bei meinen Arbeiten geholfen haben, Fragen beantworteten und allgemein für ein überaus angenehmes Arbeitsklima sorgten.
3
0. Inhaltsverzeichnis
1. Einleitung ... 7
1.1. Zielsetzung der Arbeit ... 8
2. Kenntnisstand der Literatur ... 10
2.1. Lumineszenzprozesse ... 10
2.2. Uranyl(VI)-Lumineszenz ... 12
2.3. Quenching Mechanismen des Uranyl-Chlorid-Systems ... 14
2.4. Quantifizierung des Quenchingprozesses – die Stern-Volmer-Gleichung ... 16
2.5. Komplexierungskonstanten von Uranyl(VI) mit Haliden ... 20
2.6. Detektionsmethoden zur Untersuchung des Uranyl(VI)-Quenchings ... 22
2.6.1. Untersuchung des gebildeten Intermediates - Uran(V) ... 22
2.6.2. Uran(IV) ... 26
2.6.3. Radikalfänger ... 27
3. Experimenteller Teil ... 29
3.1. Verwendete Chemikalien ... 29
3.2. Einstellung des pH-Werts ... 30
3.3. Simultane Untersuchungen der Uranyl(VI)/Uranyl(V)/Uran(IV)-Oxidationsstufen mithilfe von UV/Vis und TRLFS ... 31
3.3.1. Entwicklung eines Messaufbaus für simultane UV/Vis- und TRLFS-Experimente bei variierender Temperatur ... 31
3.3.2. Temperaturabhängige UV/Vis-Untersuchungen von Uranyl(VI)- und Uran(IV)-Lösungen ... 35
3.3.3. Untersuchungen der photoinduzierten Reduktion von Uranyl(VI) mit 2-Propanol ... 36
3.3.4. Untersuchungen der photoinduzierten Reduktion von Uranyl(VI) in Lösungen mit hoher Chloridkonzentration ... 37
3.3.5. Versuche der Stabilisierung von Uranyl(V) in Lösung mithilfe von Radikalfängern ... 38
3.4. Temperaturabhängige Untersuchungen der Uranyl(VI)-Komplexierung mit Haliden .. ... 39
3.4.1. Speziationsberechnungen der Uranyl(VI)chlorid- und Uranyl(VI)fluorid-Komplexe ... 40
3.4.2. Uranyl(VI)chlorid ... 41
3.4.3. Uranyl(VI)fluorid ... 42
3.5. Verwendete Geräte und Programme ... 42
3.5.1. Lasersystem zur Anregung von Uran(VI) ... 42
4
3.5.2. Lasersystem zur Anregung von Uran(IV) ... 43
3.5.3. UV/Vis-Geräte... 43
3.5.4. Auswertungsprogramme ... 45
4. Ergebnisse und Diskussion ... 46
4.1. Simultane Untersuchungen der Uranyl(VI)/Uranyl(V)/Uran(IV)-Oxidationsstufen mithilfe von UV/Vis-Spektroskopie und TRLFS ... 46
4.1.1. Temperaturabhängige UV/Vis-Untersuchungen von Uranyl(VI)- und Uran(IV)-Lösungen ... 46
4.1.2. Untersuchungen der photoinduzierten Reduktion von Uranyl(VI) mit 2-Propanol ... 48
4.1.3. Untersuchungen der photoinduzierten Reduktion von Uranyl(VI) in Lösungen mit hoher Chloridkonzentration ... 56
4.1.4. Versuche zur Stabilisierung von Uranyl(V) in Lösung mithilfe von Radikalfängern ... 62
4.2. Temperaturabhängige Untersuchungen der Uranyl(VI)-Komplexierung mit Haliden .. ... 67
4.2.1. Uranyl(VI)chlorid ... 67
4.2.2. Uranyl(VI)fluorid ... 83
5. Zusammenfassung und Ausblick ... 90
6. Literaturquellen ... 93
7. Selbstständigkeitserklärung ... 101
8. Anhang ... 102
5 Tabelle 1: Abkürzungsverzeichnis.
Abkürzung Bedeutung
TRLFS Engl. time resolved laser induced fluorescence spectroscopy zeitaufgelöste laserinduzierte Fluoreszenzspektroskopie
UV/Vis Engl. ultra violet/visible light
Ultraviolettes/sichtbares Licht
IR Engl. infrared radiation
Infrarote Strahlung
ISC Engl. intersystem crossing
Zwischensystemübergang
LMCT Engl. ligand to metal charge transfer
Ligand zum Metall-Ladungsübertragung HOMO Engl. highest occupied molecular orbital
Höchstbesetztes Molekülorbital
FRET Förster-Resonanz-Energietransfer
LUMO Engl. lowest unoccupied molecular orbital Niedrigstes unbesetztes Molekülorbital
DFT Engl. density functional theory
Dichtefunktionaltheorie
MCCT Engl. metal-centred charge transfer
Metallzentrierte Ladungsübertragung
DPPH Diphenylpicrylhydrazyl
XANES Engl. x-ray absorption near edge structure
Röntgenabsorptionsspektroskopie im kantennahen Bereich
NEA Nuclear Energy Agency
Kernenergie-Agentur
NIR Engl. near infrared
Nahinfrarot
CCD Engl. charge coupled devices
Ladungsgekoppeltes Bauteil
6 Tabelle 2: Formelzeichenverzeichnis.
Formelzeichen Bedeutung
KSV Stern-Volmer-Konstante
Kd Stern-Volmer-Konstante für dynamisches Quenching Ks Stern-Volmer-Konstante für statisches Quenching
log β0 Komplexierungskonstante
ε Molarer Absorptionskoeffizient
ΔrH◦m Reaktionsbildungsenthalpie
τ1/2 Halbwertszeit eines Nuklides
F0 Fluoreszenzintensität eines Fluorophors in Abwesenheit eines Quenchers F Fluoreszenzintensität eines Fluorophors in
Anwesenheit eines Quenchers
[Q] Quencherkonzentration
τ0 Fluoreszenzlebensdauer in Abwesenheit eines Quenchers τ Fluoreszenzlebensdauer in Anwesenheit eines Quenchers
kq Bimolekulare Quenchingkonstante
R universelle Gaskonstante
A Präexponentieller Faktor
Ea Aktivierungsenergie
kB Boltzmann-Konstante
h Planksches Wirkungsquantum
ΔHǂ Aktivierungsenthalpie
ΔSǂ Aktivierungsentropie
ΔGǂ Aktivierungs-Gibbs-Energie
Innerhalb der Arbeit wurde zur besseren Verständlichkeit und Lesbarkeit das angelsächsische Zahlenformat genutzt. Messungen wurden, wenn nicht anders beschrieben, bei Raumtemperatur durchgeführt.
7
1. Einleitung
Die Untersuchungen an Uran sind in Bezug auf die Nutzung der Kernenergie weltweit, sowie der Endlagerung des bereits produzierten nuklearen Abfalls in Deutschland, weiterhin von größter Wichtigkeit. Als Brennstoff in der Kernenergie wird das Element Uran genutzt. Die natürlich vorkommenden Uran-Nuklide sind sehr langlebig und zudem der Ausgang der beiden natürlichen Zerfallsreihen: Uran-235 (τ1/2 = 7.037∙108 Jahre) ist Ausgangspunkt der Uran- Actinium-Reihe und Uran-238 (τ1/2 = 4.466∙109 Jahre) der Uran-Radium-Reihe.1-2 Entsprechend ist Uran der Ursprung vieler natürlich vorkommender Radionuklide, welche Teil der natürlichen Zerfallsreihen sind. Uran-238 ist in der Natur mit einer natürlichen Häufigkeit von 99.27 % und Uran-235 mit 0.72 % anzutreffen.3 Wird der Uran-235 Anteil technisch auf einen Wert von über drei Prozent erhöht, so liegt angereichertes Uran vor.4 Zum Gewinn von Kernenergie wird angereichertes Uran genutzt, welches in Form von Brennelementen in Atomkraftwerken eingesetzt wird. Die Spaltprodukte von Uran sind über einen langen Zeitraum radiotoxisch, weshalb eine Endlagerung der Brennstäbe über mehrere tausend Jahren nötig ist.5 Dabei stellt ein Hauptteil der abgebrannten Brennelemente das nicht spaltbare Uran-238 sowie das nicht gespaltene Uran-235 dar (siehe Abbildung 1).
Entsprechend wichtig sind Messmethoden, mithilfe derer Uran gezielt untersucht werden kann, um dessen biologisches, chemisches und physikalisches Verhalten vorauszusagen.
Abbildung 1: Nuklidzusammensetzung von frischen (links) und abgebrannten (rechts) Brennelementen.6
8
Alle Konzepte für Endlager sehen vor, dass radioaktive Materialien weit unter der Erdoberfläche gelagert werden sollen. In mehreren hundert Metern Tiefe herrschen andere Temperaturbedingungen als an der Oberfläche, wobei zusätzlich ein Temperaturanstieg durch radioaktive Zerfallsprozesse zu erwarten ist.7 Somit ist es notwendig den Temperatureinfluss auf die Speziation von Uran zu evaluieren und zu analysieren, wie sich die Temperatur auf die genutzte Messmethodik auswirkt.
Eine exzellente Methode zur Untersuchung des lumineszenten Urans bietet die zeitaufgelöste laserinduzierte Fluoreszenzspektroskopie (engl. time-resolved laser induced fluorescence spectroscopy; TRLFS). Diese eignet sich in hohem Maße, um die nähere chemische Umgebung des Urans zu untersuchen.8-10 Trotz der hohen Sensitivität der Methode ist bekannt, dass die Uranlumineszenz von Haliden, ausgenommen dem Fluorid, sowie Carbonat gequencht wird.11-14 Quenching beschreibt dabei Prozesse, deren Resultat eine Verminderung der Lumineszenzintensität ist und wird auch Fluoreszenzauslöschung genannt. Daher ist eine Detektion von Uran mittels TRLFS in halidhaltigen Medien schwierig bis unmöglich und es wird oft unter Ausschluss von Haliden gearbeitet. Jedoch ist dies nicht immer praktikabel, wenn beispielsweise natürlich vorkommende Proben experimentell untersucht werden sollen, oder die Wechselwirkung zwischen Uran und biologischen Organismen näher betrachtet wird, welche Halide zum Wachstum benötigen.15-16
1.1. Zielsetzung der Arbeit
In dieser Arbeit sollte die Wechselwirkung zwischen Uran und Chlorid als quenchendes Halid sowie von Uran und Fluorid als nicht quenchendes Halid untersucht werden. Der Quenchingprozess des Urans findet photoinduziert durch den Chloridliganden statt, weshalb die TRLFS eine geeignete Methode darstellt, um den Vorgang zu untersuchen. Es ist bisher unbekannt, wie die Temperatur den Quenchingmechanismus beeinflusst. Um ein besseres Verständnis über die Art und den Ablauf des Prozesses zu gewinnen, wurden in der Arbeit Untersuchungen bei variierenden Temperaturen durchgeführt.
Zum Zweck der Untersuchung ist es notwendig eine geeignete Methode zu entwickeln, welche es erlaubt, Uran bei hohen Chloridkonzentrationen zu untersuchen und eine Speziation des Urans mit Chlorid nach Laserbestrahlung zu detektieren. Optische Methoden, welche sich
9
hierfür anbieten, sind die UV/Vis- (engl. Ultraviolet/visible) und die IR- (engl. Infrared) Absorptionsspektroskopie. Dabei ist es notwendig, die Absorptionsmessungen simultan zur Laseranregung der TRLFS durchzuführen, um den Quenchingprozess exakt beobachten zu können. Aus diesem Grund ist auch die Methodenentwicklung zur Simultanmessung von IR-/UV/Vis-Absorption und TRLFS Teil dieser Arbeit.
10
2. Kenntnisstand der Literatur
2.1. Lumineszenzprozesse
Um den Vorgang des photoinduzierten Quenchings mithilfe der TRLFS beschreiben zu können, ist es notwendig, die grundlegenden Prozesse der Lumineszenz zu erläutern.
Ein Elektron kann durch die Absorption von Energie in Form von elektromagnetischer Strahlung, einem Photon, aus seinem energetischen Grundzustand in einen energetisch angeregten Zustand übergehen. Der Absorptionsbereich von Elektronen für elektromagnetische Strahlung erstreckt sich hierbei je nach Atom- und Molekülstruktur über den UV- bis in den Vis-Bereich. Indem die Probe mit einem kontinuierlichen Spektrum bestrahlt wird, kann die Absorption von elektromagnetischer Strahlung über Absorptionsspektroskopie nachgewiesen werden. Mittels eines Detektors wird daraufhin nachgewiesen, welche Wellenlängenbereiche von der Probe absorbiert wurden.17
Der Übergang der Elektronen durch die Absorption von elektromagnetischer Strahlung vollzieht sich dabei nicht direkt vom energetischen Grundzustand in den energetisch angeregten Zustand. Eine Anregung findet nach dem Franck-Condon-Prinzip zuerst in einen energetisch angeregten vibronischen Zustand statt. Dies ist darauf zurückzuführen, dass Elektronen eine gewisse Aufenthaltswahrscheinlichkeit in jedem vibronischen Zustand aufweisen. Diese Aufenthaltswahrscheinlichkeiten werden quantenmechanisch durch Wellenfunktionen auf einer Potentialkurve beschrieben (siehe Abbildung 2).18
Dabei ist ein Übergang eines Elektrons am wahrscheinlichsten, wenn die Maxima zweier Wellenfunktionen ähnliche nukleare Koordinaten besitzen. Da sich durch eine Anregung eines Elektrons jedoch die molekulare Struktur verändert, ändert sich ebenso die Lage der Potentialkurve im angeregten Zustand. Somit ist ein Übergang vom Maximum des elektronischen Grundzustands in das Maximum eines vibronisch angeregten Zustandes am wahrscheinlichsten.
11
Abbildung 2: Schematische Darstellung des Franck-Condon-Prinzipes.19
Da die Elektronen immer einen energetisch niedrigen Zustand anstreben, gehen diese zunächst strahlungslos durch Schwingungen in den ersten angeregten energetischen Zustand über. Von diesem Niveau aus können sie nach einer begrenzten Aufenthaltsdauer in ein vibronisches Niveau, welches sich energetisch über dem Grundzustand befindet, oder dem Grundzustand übergehen.
Abbildung 3: Franck-Condon-Prinzip mit der Darstellung eines intersystem crossings (ISC) und folgender Phosphoreszenz.20
Auch hier ist der Übergang wahrscheinlicher, wenn die Lage der Maxima beider Wellenfunktionen sich ähnelt. Falls der energetische Abstand zwischen den beiden Zuständen groß genug ist, erfolgt die Depopulation unter Emission von elektromagnetischer Strahlung.
12
Dieser Prozess wird als Fluoreszenz bezeichnet. Das Photon kann hierbei durch verschiedene Messmethodiken detektiert werden.17, 19 Das Elektron befindet sich daraufhin in einem vibronischen Niveau, von welchen es erneut strahlungslos in den elektronischen Grundzustand übergehen kann.
Alternativ ist es dem Elektron möglich, ein intersystem-crossing aus dem angeregten elektronischen Zustand zu vollziehen. Durch diesen „verbotenen“ (unwahrscheinlichen) Spinwechsel geht das Elektron hierbei von einem Singulett- in einem Triplett-Zustand über, welcher sich unterhalb des elektronisch angeregten Singulett-Zustands befindet.
Veranschaulichend wird dies als eine weitere Potentialkurve dargestellt (siehe Abbildung 3).
Wenn das Elektron nun in den Grundzustand übergehen soll, muss es zuerst einen weiteren Spinwechsel vollziehen. Denn nach dem Pauli-Prinzip dürfen sich in einem energetischen Zustand, einem Orbital, nur zwei Elektronen mit jeweils gegensätzlichen Spin aufhalten. Da die Wahrscheinlichkeit für eine Spinumkehr gering ist, ist die Phosphorenz auf einer zeitlichen Skala länger als die Fluoreszenz.21
2.2. Uranyl(VI)-Lumineszenz
Uran besitzt die Elektronenkonfiguration [Rn] 5f3 6d1 7s2. Dies bedeutet es besitzt insgesamt sechs Außenelektronen. In der Natur kommt Uran unter oxischen Bedingungen meist in der Oxidationsstufe U6+ vor (siehe Abbildung 4).22
Abbildung 4: Pourbaix-Diagramm für eine Urankonzentration von 1 M. Die gestrichelten Linien zeigen den Stabilitätsbereich von Wasser an.23
13
Das bedeutet es sollte keine Lumineszenz des Urans möglich sein, da das Uran keine Elektronen besitzt, welche angeregt werden können. Jedoch bildet das hochgeladene Uran(VI)-Kation in Gegenwart von Sauerstoff das Uranyl(VI) mit der Strukturformel UO22+.22 Das Uran-Atom ist im UO22+ kovalent mit zwei Sauerstoffatomen verbunden, welche ein lineares Molekül ausbilden. Bereits in leicht sauren Milieus findet eine weitere Hydrolyse des Uranyl(VI) statt, indem äquatoriale Wassermoleküle dissoziieren und somit Uranyl(VI)hydroxide gebildet werden.8, 24 Durch Bestrahlung mit Licht findet im Uranyl(VI) ein Ladungsübertrag vom Ligand zum Metall statt (LMCT; engl. ligand to metal charge transfer).25 Der Sauerstoff fungiert hierbei als Elektronendonor für das Urankation, wodurch das Uran anregbare Elektronen in Form der Uran-Sauerstoff-Bindung besitzt. Durch Anregung dieser Bindungselektronen ist es möglich, die Lumineszenz des Uranyl(VI) zu beobachten. Die Lumineszenz ist charakteristisch geprägt durch die Aufspaltung der Bande in sechs einzelne
„Finger“, welche Fluoreszenzbanden darstellen. Die Banden befinden sich in einem Abstand von etwa 855 cm-1 zueinander. Die 855 cm-1 entsprechen der Frequenz der symmetrischen Schwingung des Grundzustandes.26-28
Abbildung 5: Ausschnitt des Zusammenhangs zwischen der Aufspaltung der Uranyl(VI)-Lumineszenz in einzelne Banden und der Schwingung des Uranyl(VI)-Grundkörpers. 29
Entsprechend kann ein Elektron aus dem angeregten Zustand in die fünf vibronischen Niveaus des Grundzustandes übergehen oder eine Depopulation aus einem vibronischen Level des energetisch angeregten Zustandes erfolgen (siehe Abbildung 5).30 Dies führt zu einer Aufspaltung der Lumineszenz in insgesamt sechs Banden. Anhand der Lage und der
14
Intensitätsverhältnisse der Banden zueinander ist es möglich, die chemische Umgebung des Uranyl(VI) zu analysieren und eine mögliche Speziation festzustellen.31-33
2.3. Quenching Mechanismen des Uranyl-Chlorid-Systems
Die Lumineszenz des Urans wird in der Gegenwart von Haliden mit der Ausnahme von Fluorid gequencht, was zu einer Verminderung der Fluoreszenzintensität führt. In der Literatur werden verschiedene Theorien beschrieben, wie der Quenchmechanismus des Urans mit Haliden stattfindet. Yokoyama et. al (1976) beschreiben einen Mechanismus, bei welchen ein Elektronentransfer durch Quenching via Kollision stattfindet.11 Dabei wird ein Lumineszer, in diesem Fall das Uranyl(VI), angeregt, welcher mit einem Quencher, dem Halid, ein Kollisionskomplex bildet. Im Uranyl(VI) befindet sich das anzuregende Elektron im höchst besetzten Molekülorbital (engl. highest occupied molecular orbital; HOMO), welches im Uranyl(VI) ein π-Orbital ist. Durch die Kollisionskomplexbildung kommt es zu einer Übertragung des Elektrons vom Quencher zum Lumineszer. Das Elektron wird vom HOMO des Uranyl(VI) in das niedrigste unbesetzte Molekülorbital (engl. lowest unoccupied molecular orbital; LUMO) angeregt. Das LUMO ist hierbei ein 5f-Orbital. Im HOMO des Uranyl(VI) entsteht daraufhin eine Lücke, welche durch ein Elektron vom HOMO des Haliden gefüllt wird.
Die entstehende Lücke im HOMO des Haliden wird durch das Elektron aus dem LUMO des zwischenzeitlich gebildeten Uranyl(V) gefüllt. Dadurch entstehen wieder Uranyl(VI) und Halid im jeweiligen elektronischen Grundzustand und es erfolgt eine Auflösung des Kollisionskomplexes. Der stattfindende Prozess muss dabei thermodynamisch erlaubt sein, damit ein Quenching stattfindet. Das Quenching von Uranyl(VI) mit Chlorid, Bromid und Iodid ist möglich, wobei energetisch der Quenchingprozess vom Chlorid zum Iodid zunehmend freiwilliger wird und somit Iodid am stärksten die Fluoreszenz abschwächt. Hingegen ist das Quenching mit Fluorid energetisch nicht freiwillig, weshalb keine Abschwächung der Uranfluoreszenz mit Fluorid erfolgt.
Hingegen nimmt Burrows (1990) an, dass das Quenching von Uranyl(VI) in Zusammenhang mit der Bildung von Dihalidradikalanionen steht.14 Dies würde vor allem für Bromid, Iodid als auch das Pseudohalogenid Thiocyanat zutreffen. Auch mit Chlorid soll eine Bildung eines Dihalidradikalanions möglich sein, jedoch spielt dieses im Quenchingprozess des
15
Chlorid-Uranyl(VI)-Systems nur eine untergeordnete Rolle. Burrows beschreibt eine Bildung eines Exciplexes zwischen dem angeregten Uranyl(VI) und dem Halidanion (1). Der Exciplex zerfällt wieder in das Uranyl(VI) und das Halidanion im elektronischen Grundzustand (2) oder reagiert mit einem weiteren Halidanion zum Uranyl(V) und dem Dihalidradikalanion (3).
∗𝑈𝑂
22++ 𝑋−→ (𝑈𝑂2+𝑋∙) (1) (𝑈𝑂2+𝑋∙) → 𝑈𝑂∗ 22++ 𝑋− (2)
(𝑈𝑂2+𝑋∙) + 𝑋−→ 𝑈𝑂2++ 𝑋2∙− (3)
Beide beschriebenen Theorien nehmen an, dass eine Abnahme der Uranyl(VI)-Lumineszenz durch die Bildung von Uranyl(V) stattfindet. Entsprechen könnten beide Methoden bekräftigt werden, wenn eine Detektion von Uranyl(V) oder dem durch Disproportionierung entstehenden Uran(IV) möglich wäre.
Tsushima et. al (2010) versuchten unter anderem mit quantenchemischen Berechnungen den Quenchingprozess von Burrows nachzuvollziehen.13 Mithilfe von DFT-Berechnungen studierten sie hierbei die UO2X(H2O)4+ Komplexe (X=Halide). Sie untersuchten die Anregungsenergie, welche nötig ist, damit ein Elektron von den bindenden Orbitalen (σu, σg, πg, πu) in die nicht bindenden Orbitale (5fδ, 5fφ) übergeht. Dabei bilden sich sowohl Singulett- als auch Triplettelektronenzustände, wobei der Triplettzustand energetisch zumeist niedriger liegt und somit die Fluoreszenz (Phosphoreszenz) möglich ist. In der siebten Hauptgruppe ist hierbei die vertikale Übergangsenergie für den Komplex mit Fluorid am größten, während die Energie für Iodid am kleinsten ist. Aus diesem Grund ist die Fluoreszenz des Uranyls mit Fluorid sichtbar, während das Quenching mit steigender Periode in der Hauptgruppe zum Iodid zunimmt. Berechnungen des Bindungsabstandes zwischen Uranatom und Halid zeigen auf, dass der Abstand von Fluorid zum Iodid hin steigt. Uran-Fluorid befinden sich in einem Komplex, während Iodid sich so weit vom Uranatom entfernt hat, dass es möglich ist, von einem dissoziierten Komplex zu sprechen. Über eine Berechnung der Elektronenspindichte schlussfolgerten Tsushima et. al, dass Fluorid kein Quencher ist und Iodid am stärksten quencht, während sich Chlorid und Bromid dazwischen ansiedeln. Aus der energetischen molekularen Lokalisation der HOMOs und LUMOs in den verschiedenen Komplexen, war es
16
ihnen möglich vorauszusagen, dass im UO2F+ ein metallzentrierter Ladungstransfer (engl.
metal-centered charge transfer; MCCT) und im UO2I+ ein LMCT stattfindet. UO2Cl+und UO2Br+ wiederum befinden sich von ihrem Charakter zwischen MCCT und LMCT, werden jedoch tendenziell eher einem LMCT zugeordnet. Als LMCT-Komplex liegt dabei UO2I+ als (𝑈𝑂2+𝑋∙) vor, was die Theorie von Burrows bekräftigen würde.
Uranyl(VI) wird jedoch nicht nur durch die Anwesenheit von Haliden gequencht, sondern auch durch die Komplexbildung mit Wasser zum [UO2(H2O)5]2+. Formosinho et. al (2003) wiesen darauf hin, dass die kurze Lebensdauer von 1.9 μs des Uranyl(VI) bei pH < 1.5 auf Quenching durch Wasserstoffabstraktion zurückzuführen ist.34 Die Lebensdauer des Uranyl(VI) ist zu kurz, als dass das Quenching nur durch Schwingungen des Uranyl-Aquo-Komplexes stattfinden würde. Bei dieser Reaktion würde das Uranyl(V) entstehen:
∗𝑈𝑂
22++ 𝐻2𝑂 → 𝑈𝑂2𝐻2++ 𝑂𝐻∙ (4)
𝑈𝑂2𝐻2+→ 𝑈𝑂2++ 𝐻+ (5)
Es ist bekannt, dass Uran(V) instabil ist und eine Tendenz zu Disproportionierung aufweist und in U(VI) sowie U(IV) zerfällt.35 Die Detektion von Uran(V) würde einen eindeutigen Hinweis auf die postulierten Quenchingmechanismen liefern. Jedoch wird Uranyl(V) laut Formosinho et. al auch sehr schnell über Hydroxyradikale zurück zu Uranyl(VI) oxidiert:
𝑈𝑂2++ 𝑂𝐻∙ → 𝑈𝑂22++ 𝑂𝐻− (6)
Dies erschwert die Detektion von Uran(V), da Hydroxyradikale durch das Quenching von Wasser mit Uranyl(VI) in Lösung immer vorhanden sind, wenn eine energetische Anregung mittels Licht erfolgt. Der Einfluss von Sauerstoff auf die Rückoxidation wird hingegen von Formosinho et. al als gering und langsamer Prozess gewertet.
2.4. Quantifizierung des Quenchingprozesses – die Stern-Volmer-Gleichung
Eine bekannte Theorie um Quenchingprozesse zu beschreiben und zu quantifizieren ist der Förster-Resonanzenergietransfer (FRET).17 Im Falle eines FRET wird die Energie eines angeregten Moleküls, dem Donor, an einen Akzeptor übertragen.36 Im Donor erfolgt eine
17
Depopulation des angeregten Elektrons unter Emission von elektromagnetischer Strahlung.
Die Strahlung wird wiederum von dem Elektron des Akzeptors im Grundzustand absorbiert, welches dadurch angeregt wird. Das angeregte Elektron des Akzeptors wiederum depopuliert strahlungslos oder unter Abgabe von langwelligerer Strahlung als das Elektron des Donors.
Entsprechend wird die Lumineszenz des Donors gequencht. Jedoch wird in den unter 2.3 beschriebenen Theorien kein Energietransfer beschrieben, sondern eine Desaktivierung durch Elektronenübergang. Entsprechen muss eine andere Methodik zur Quantifizierung der Fluoreszenzauslöschung genutzt werden.
Eine quanti- und qualitative Analyse eines Quenchingprozesses ist mithilfe der Stern-Volmer- Gleichung möglich (siehe (7)). Diese Gleichung erlaubt eine Auftragung der Fluoreszenzintensität des Fluorophors in Abwesenheit eines Quenchers F0 geteilt durch die Fluoreszenzintensität des Fluorophors in Anwesenheit eines Quenchers F gegen die Konzentration des Quenchers [Q].17, 37
𝐹0
𝐹 − 1 = 𝐾𝑆𝑉∙ [𝑄] (7)
Aus dem gebildeten linearen Verhalten ist es möglich, den Anstieg einer Gerade zu erhalten.
Dieser Anstieg ist die Stern-Volmer-Konstante KSV. Die Stern-Volmer-Konstante wird je nach vorliegendem Quenchingprozess verschieden beschrieben.
Im Falle des dynamischen Quenchings ergibt sich der Zusammenhang für die Konstante aus der Lebensdauer des Fluorophores in Abwesenheit eines Quenchers τ0 und einer bimolekularen Quenchingkonstante kq (siehe (8)).
𝐾𝑆𝑉= 𝜏0∙ 𝑘𝑞 (8)
Dynamisches Quenching beschreibt einen Prozess, bei dem die Energie des angeregten Fluorophores durch eine bimolekulare Kollision an einen Quencher abgegeben wird. Somit erfolgt ein strahlungsloser Rückgang des angeregten Fluorophors in den energetischen Grundzustand. Bei konstantem τ0 ergibt sich, dass mit steigendem kq eine Kollision zweier
18
Teilchen wahrscheinlicher wird und somit die Effizienz des Quenchings steigt. Für das dynamische Quenching ergibt sich zudem folgende Gleichung:17
𝐹0
𝐹 − 1 =𝜏0
𝜏 − 1 = 𝐾𝑆𝑉∙ [𝑄] (9)
Aus (9) ergibt sich, dass das Verhältnis zwischen ungequenchter und gequenchter Intensität gleich dem Verhältnis der ungequenchten zur gequenchten Lebensdauer ist. Somit sollte eine Auftragung beider Größen gegen die Quencherkonzentration den gleichen Wert für KSV liefern (siehe Abbildung 6). Dies ergibt sich aus dem Zusammenhang, dass eine Kollision zwischen einem angeregten Fluorophor und einem Quencher beim dynamischen Quenching umso wahrscheinlicher wird, desto länger die Lebensdauer ist. Mit steigender Temperatur sollte zudem KSV zunehmen, da durch die erhöhte Brownsche Molekularbewegung eine Kollision wahrscheinlicher wird.
Abbildung 6: Schematische Darstellung zum Verhältnis von F0/F bzw. τ0/τ gegen die Quencherkonzentration [Q]
im Falle eines dynamischen (links) und eines statischen Quenchingprozesses (rechts).38
Eine weitere Form des Quenchings ist das statische Quenching. In dem Falle eines rein statischen Quenchings entspricht dabei KSV der Komplexbildungskonstante nach dem Masse- Wirkungs-Gesetz, da ein nicht anregbarer Komplex gebildet wird (siehe (10)).
𝐾𝑆𝑉= [𝐹𝑄]
[𝐹] ∙ [𝑄]
(10)
Im Vergleich zum dynamischen Quenching ist das Verhältnis von Lebensdauer in Abwesenheit eines Quenchers zur Lebensdauer in Anwesenheit eines Quenchers immer konstant (siehe (11) und Abbildung 6).
19 𝜏0
𝜏 = 1 (11)
Statisches Quenching vermindert lediglich die Konzentration des nicht angeregten Fluorophors, allerdings nicht die Lebensdauer des Fluorophors sobald es angeregt ist. Der Einfluss der Temperatur auf das Quenching hängt hierbei davon ab, ob eine erhöhte Temperatur eine Bildung des quenchenden Komplexes fördert oder nicht.
Dynamisches und statisches Quenching können zudem in Kombination miteinander auftreten (siehe Abbildung 7).
Abbildung 7: Schematische Darstellung des Unterschiedes zwischen dynamischen Quenching und einer Kombination aus dynamischen und statischen Quenching.39
Um den Zusammenhang zwischen dynamischen und statischen Quenching beschreiben zu können, muss die Stern-Volmer-Gleichung modifiziert werden. Hierzu wird zwischen der Stern-Volmer-Konstante für dynamisches Quenching Kd und statisches Quenching Ks
unterschieden (siehe (12)).17 𝐹0
𝐹 = (1 + 𝐾𝑑∙ [𝑄]) ∙ (1 + 𝐾𝑠∙ [𝑄]) (12)
(12) vereinfacht sich durch (9) zu (13).
𝐹0 𝐹 =𝜏0
𝜏 ∙ (1 + 𝐾𝑠∙ [𝑄]) (13)
Aus (13) ergibt sich, dass das Verhältnis der Lebensdauern 𝜏𝜏0 zueinander kleiner sein muss als das Verhältnis der Fluoreszenzintensitäten 𝐹0
𝐹 zueinander.
20
2.5. Komplexierungskonstanten von Uranyl(VI) mit Haliden
Zum Zweck der Ermittlung der experimentellen Konditionen sind Literaturwerte für die Bildung der verschiedenen Uranyl(VI)-Komplexe mit Haliden notwendig.
Tabelle 3: Bekannte Komplexierungskonstanten log β0 für Uranyl(VI)fluorid-Komplexe.
Spezies Reaktion log β0 Methode Ionenstärke [F] Temperatur Literatur
UO2F+ UO22+ + F- ↔ UO2F+
5,15±0,08 Chinhydron- elektrode
1 M NaClO4 0,01- 0,025 M
20 °C Ahrland et. al40
5,25±0,10 NMR 0,5 M
NaClO4
- 25 °C Vdovenko et. al41 5,14±0,08 Chinhydron-
elektrode;
Ionenselektive Elektrode
1 M NaClO4 0,0006- 0,4 M
25 °C Ahrland et. al42
5,13±0,09 Ionenselektive
Elektrode 1 M NaCl 0,00001- 0,1 M
25 °C Ishiguro et. al43
5.1 Berechnung - - 25 °C Langmuir et. al44
5,1±0,3 Berechnung - - 25 °C Lemire et. al45
5,17±0,08 Ionenselektive
Elektrode 1 M NaClO4 0,004- 0,156 M
21 °C Sawant et. al46 UO2F2 UO22+ + 2 HF ↔
UO2F2 + 2 H+
8,3±0,2 Kationen- austausch
0,2 M HClO4 - 25 °C Krylov et. al47 UO22+ + 2 F- ↔
UO2F2
8,56±0,07 Chinhydron-
elektrode 1 M NaClO4 0,01- 0,025 M
20 °C Ahrland et. al40 8,64±0,07 Chinhydron-
elektrode;
Ionenselektive Elektrode
1 M NaClO4 0,0006- 0,4 M
25 °C Ahrland et. al42
8,60±0,08 Ionenselektive
Elektrode 1 M NaCl 0,00001- 0,1 M
25 °C Ishiguro et. al43
9 Berechnung - - 25 °C Langmuir et. al44
UO2F3- UO22+ + 3 HF ↔ UO2F3- + 3 H+
11.2 Kationen- austausch
2 M HClO4 - 25 °C Krylov et. al47
UO22+ + 3 F- ↔ UO2F3-
11,09±0,10 Chinhydron-
elektrode 1 M NaClO4 0,01- 0,025 M
20 °C Ahrland et. al40 11,04±0,10 Chinhydron-
elektrode;
Ionenselektive Elektrode
1 M NaClO4 0,0006- 0,4 M
25 °C Ahrland et. al42
10,64±0,11 Ionenselektive
Elektrode 1 M NaCl 0,00001- 0,1 M
25 °C Ishiguro et. al43
11.3 Berechnung - - 25 °C Langmuir et. al44
11,3±0,4 Berechnung - - 25 °C Lemire et. al45
5,17±0,08 Ionenselektive
Elektrode 1 M NaClO4 0,004- 0,156 M
21 °C Sawant et. al46 UO2F42- UO22+ + 4 F- ↔
UO2F42-
11,90±0,12 Chinhydron- elektrode
1 M NaClO4 0,01- 0,025 M
20 °C Ahrland et. al40 11,98±0,12 Chinhydron-
elektrode;
Ionenselektive Elektrode
1 M NaClO4 0,0006- 0,4 M
25 °C Ahrland et. al42
11,11±0,15 Ionenselektive
Elektrode 1 M NaCl 0,00001 -0,1 M
25 °C Ishiguro et. al43
12.6 Berechnung - - 25 °C Langmuir et. al44
12,6±0,4 Berechnung - - 25 °C Lemire et. al45
21
Die Literaturwerte sowie die Literaturquellen wurden dabei der NEA-Datenbank (Nuclear Energy Agency) für Uran entnommen.48 Weiterhin geben Komplexbildungskonstanten Aufschluss darüber, ob statisches oder dynamisches Quenching möglich ist. Eine Bildung eines Komplexes würde statisches Quenching implizieren, was zu einer Reduktion an Lumineszenzintensität führt. Falls die Bildung eines Komplexes unwahrscheinlich ist, kann von rein dynamischen Quenching ausgegangen werden.
Dabei werden nur die Daten für die Uranyl(VI)-Komplexe mit Fluorid, Chlorid und Bromid dargestellt, da keine Daten für die Bildung eines Uranyl(VI)iodid-Komplex vorliegen, vermutlich aufgrund der großen Instabilität eines solchen Komplexes. Die thermodynamischen Größen der Komplexierung von Uranyl(VI) mit Fluorid wurden in der Literatur sehr extensiv untersucht, wie an Tabelle 3 zu erkennen ist. Anhand der Komplexierungskonstanten log β0 lässt sich abschätzen, dass die Uranyl(VI)fluorid-Komplexe zudem weitgehend stabil sind.
Tabelle 4: Bekannte Komplexierungskonstanten log β0 für Uranyl(VI)chlorid-Komplexe.
Spezies Reaktion log β0 Methode Ionenstärke [Cl] Temperatur Literatur
UO2Cl+ UO22+ + Cl- ↔ UO2Cl+
0.38 Spektro-
photometrie Elektromotor- ische Kraft
variierend 0,0075-2 M 25 °C Nelson et. al49
0,22±0,03 Spektro-
photometrie 0,05-0,12 M Na(Cl;ClO4)
0,01-3 M 25 °C Bale et. al50 1.29 Elektromotor-
ische Kraft
0,05-0,5 M - 15 °C Ohashi et. al51
1.19 Elektromotor-
ische Kraft 0,05-0,5 M - 20 °C
1.11 Elektromotor-
ische Kraft 0,05-0,5 M - 25 °C
0.2 Berechnung - - 25 °C Langmuir et.
al44
2,0±1,0 Berechnung - - 25 °C Lemire et. al45
0,23±0,02 Elektromotor-
ische Kraft variierend 0,1-4,8 M 25 °C Awasthi et. al52 0,27±0,02 Elektromotor-
ische Kraft variierend 0,1-4,8 M 35 °C UO2Cl2 UO22+ + 2 Cl-
↔ UO2Cl2
-1,20±0,03 Spektro-
photometrie variierend 0,1-4,8 M 25 °C -1,10±0,03 Spektro-
photometrie variierend 0,1-4,8 M 35 °C -1,00±0,03 Spektro-
photometrie variierend 0,1-4,8 M 45 °C
Ein anderes Bild hingegen liefern die Stabilitätskonstanten für die Uranyl(VI)chlorid-Komplexe, welche wesentlich kleiner sind als die der Fluorid-Komplexe (siehe Tabelle 4). Dies bedeutet,
22
dass die Chlorid-Komplexe instabiler sind und sich erst bei sehr hoher Chloridkonzentration bilden. Daher ist es auch schwierig Komplexe mit mehr als zwei Chloridliganden zu beobachten, da vorher eine Absättigung der Lösung erfolgt.
Dem Trend der Hauptgruppe folgend sind die Uranyl(VI)bromid-Komplexe noch instabiler als die Uranyl(VI)chlorid-Komplexe und somit sind die Werte der Stabilitätskonstanten in Tabelle
5 noch kleiner als für die anderen Halide. Entsprechend war es bisher auch nur möglich, den eins zu eins Komplex für das Uranyl(VI)bromid zu beobachten. Analog zur größer werdenden Instabilität der Komplexe in der siebten Hauptgruppe nimmt auch der Quenchingeffekt zu.
Aus diesem Grund wurden in dieser Arbeit keine Bromidproben untersucht, da die Hauptuntersuchungsmethode die Laserfluoreszenzspektroskopie ist und eine Detektion der Komplexe extrem schwierig wäre.
Tabelle 5: Bekannte Komplexierungskonstanten log β0 für Uranyl(VI)bromid-Komplexe.
Spezies Reaktion log β0 Methode Ionenstärke [Br] Temperatur Literatur
UO2Br+ UO22+ + Br- ↔ UO2Br+
0,24±0,17 Elektromotor- ische Kraft
1 M Na(Br;ClO4)
0,2-0,7 M 20 °C Ahrland et. al53 0,20±0,05 Spektro-
photometrie
0,532 M Na(Br;ClO4)
0,2-0,8 M 25 °C Davies et. al54 0,19±0,04 Spektro-
photometrie
0,367 M Na(Br;ClO4)
0,2-0,8 M 25 °C 0,24±0,04 Spektro-
photometrie
0,317 M Na(Br;ClO4)
0,2-0,8 M 25 °C 0,26±0,04 Spektro-
photometrie
0,217 M Na(Br;ClO4)
0,2-0,8 M 25 °C 0,19±0,04 Spektro-
photometrie
0,168 M Na(Br;ClO4)
0,2-0,8 M 25 °C
2.6. Detektionsmethoden zur Untersuchung des Uranyl(VI)-Quenchings 2.6.1. Untersuchung des gebildeten Intermediates - Uran(V)
Die unter 2.3 beschriebenen Theorien gehen davon aus, dass sich im Zuge des Quenchingprozesses von Uranyl(VI) mit Chlorid das Uranyl(V) bildet. Entsprechend würde eine Beobachtung des Uranyl(V) postulierte Theorien bestätigen.
Die Instabilität von Uran(V) erschwert jedoch die Detektion des Oxidationszustands mithilfe verschiedener Messmethoden (siehe auch Abbildung 4).27 Die häufigste genutzte optische Messmethode stellt die UV/Vis-Absorption dar. Dennoch ist die existierende Literatur in Bezug auf Uran(V) eher spärlich. Ogura et. al (2011) wiesen Uran(V) in einer ionischen Flüssigkeit,
23
welche aus 1-Ethyl-3-methylimidazoliumchlorid und 1-Ethyl-3-methylimidazoliumtetra- fluoroborat in einer 50:50 Mischung bestand, bei Raumtemperatur nach.55 Über Cyclovoltammetrie Experimente waren sie in der Lage, UVIO2Cl42- zu UVO2Cl43- zu oxidieren.
Neben dem typischen UVIO2Cl42- Signal bei 430 nm konnten sie drei neue Banden beobachten, welche sich mit verändernden Potential ausbildeten. Eine intensive Bande konnte bei einer Wellenlänge von 406 nm und zwei schwächere Banden bei Wellenlängen von 630 und 770 nm beobachtet werden (siehe Abbildung 8).
Abbildung 8: Beobachtete UV/Vis-Banden einer U(V)-Spezies von a) Ogura et. al bei 406; 630 und 770 nm und b) Salomone et. al bei 736; 845 und 962 nm.55-56
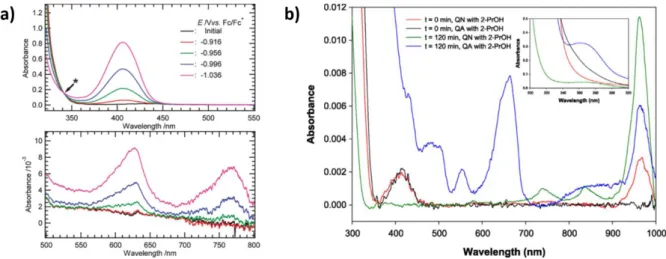
Salomone et. al (2014) führten eine optisch induzierte Reduktion von Uranyl(VI) mithilfe von Acetat durch.56 Bei einem pH von 3 und einer Konzentration von 0.25 mM Uranyl(VI) wurde dieses zu Uranyl(V) reduziert. In Folge dessen konnte eine Abnahme des Uranyl(VI)-Signals von 350-500 nm über die Zeit beobachtet werden, während eine Bande mit einer hohen Intensität bei 962 nm und zwei Banden mit geringerer Intensität bei 736 nm und 845 nm erschienen (siehe Abbildung 8). Aufgrund des Stabilitätsbereichs von Uranyl(V) zwischen pH 2-3 wurden die Signale dem Uranyl(V) zugeordnet. Weiterhin haben Salomone et. al (2015) eine Versuchsreihe mit 2-Propanol durchgeführt, welchem eine Uranyl(VI)-Lösung unter gleichen Bedingungen zugesetzt wurde.56-57 Durch optische Anregung wird das 2-Propanol zu Aceton oxidiert und Uranyl(VI) zu Uranyl(V) reduziert. Auch an dieser Stelle konnten intensivere Banden bei 737, 845 und 963 nm beobachtet werden, sowie ein Plateau zwischen 240-270 nm. Zudem führte die Bestrahlung über 2 h zur Entstehung von Uran(IV)-Banden bei 435; 490; 554 und 663 nm.
24
Auch im IR-Bereich wurden Versuche unternommen U(V) zu analysieren. Cohen et. al (1970) untersuchten die Reduktion von U(VI) zu U(V) und U(IV) in gesättigter CaCl2-Lösung mit D2O als Lösungsmittel.58 Mit zunehmender elektrolytischer Reduktion konnten sie die Entstehung von Absorptionsbanden bei 760 und 960 nm beobachten, sowie ein intensives Absorptionsmaximum um 1510 nm, welches zuvor schon von Adams et. al (1963) dem UO2+
in Chloridsalzschmelzen zugeordnet wurde.59 In nicht deuterierter 1 – 3 M K2CO3-Lösung bei pH > 11 beobachteten Cohen et. al hingegen die Entstehung von Absorptionsbanden bei 765, 980 und 1120 nm.58 Über Rücktitration mit Eisenionen bestätigten sie, dass die Banden ebenso durch das U(V) hervorgerufen werden. Die Unterschiede in den Lösungen lies die Autoren darauf schließen, dass das U(V) in beiden Lösung in einer unterschiedlichen Speziation vorliegt. Die molaren Absorptivitäten sind jedoch mit 2-3 M-1 cm-1 relativ gering. Die Resultate von Cohen wurden von Mizuoka et. al (2005) bestätigt, welche zusätzliche Absorptionsbanden bei 1600 und 1800 nm identifizieren konnten, die auch eine molare Absorptivität von 0.2- 3.6 M-1 cm-1 aufwiesen.60
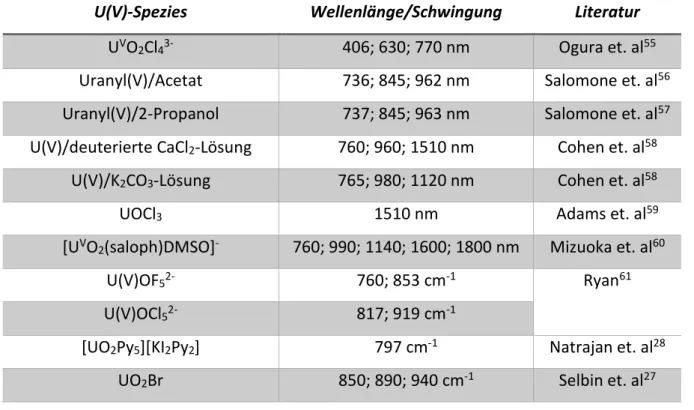
Tabelle 6: Tabellierte Werte der UV/Vis-/IR-Banden verschiedener U(V)-Spezies.
U(V)-Spezies Wellenlänge/Schwingung Literatur UVO2Cl43- 406; 630; 770 nm Ogura et. al55 Uranyl(V)/Acetat 736; 845; 962 nm Salomone et. al56 Uranyl(V)/2-Propanol 737; 845; 963 nm Salomone et. al57 U(V)/deuterierte CaCl2-Lösung 760; 960; 1510 nm Cohen et. al58
U(V)/K2CO3-Lösung 765; 980; 1120 nm Cohen et. al58
UOCl3 1510 nm Adams et. al59
[UVO2(saloph)DMSO]- 760; 990; 1140; 1600; 1800 nm Mizuoka et. al60
U(V)OF52- 760; 853 cm-1 Ryan61
U(V)OCl52- 817; 919 cm-1
[UO2Py5][KI2Py2] 797 cm-1 Natrajan et. al28
UO2Br 850; 890; 940 cm-1 Selbin et. al27
25
Weiterhin ist es möglich, die Schwingungsbanden von Uranyl(V) nachzuweisen. Die charakteristischen Uran-Sauerstoff-Schwingungen wurden in Salzen von U(V)OF52- mit Tetraethylammonium als Gegenion bei 760 und 853 cm-1 und in U(V)OCl52- bei 817 und 919 cm-1 von Ryan (1971) bestimmt.61 Natrajan et. al (2006) untersuchten U(VI) und U(V) mit organischen Pyridin-Liganden und stellten fest, dass die Länge der Bindung zwischen Uran und Sauerstoff mit der Abnahme des Oxidationszustandes des Urans steigt.28 Damit einhergehend findet eine Verschiebung der Streckschwingung von 927 cm-1 des U(VI) zu 797 cm-1 des U(V) statt. Dies ist ein Resultat der höheren Elektronendichte am Uran-Atom, was zu einer Abschwächung der Uran-Sauerstoff-Bindung führt. Während der Untersuchung von UO2Br ordneten Selbin et. al (1969) die Banden um 940, 890 und 850 cm-1 den U(V)−O Schwingungen zu.27 Eine Übersicht aller UV/Vis-Banden und IR-Schwingungsbanden ist in Tabelle 6 dargestellt.
Steudtner et. al (2006) nutzten ebenfalls die Reduktion von Uranyl(VI) mit 2-Propanol um Uranyl(V) mithilfe der TRLFS nachzuweisen.62 Durch eine Quecksilberdampflampe wurde die optisch-induzierte Reduktion mit 2-Propanol durchgeführt, woraufhin sowohl TRLFS- als auch UV/Vis-Messungen durchgeführt wurden. Der pH-Bereich betrug 2-3, in welchem Uranyl(V) ca. eine Stunde stabil sein soll.63 Im Absorptionsspektrum wurde eine Absorptionsbande der Wellenlänge von 255 nm, welche den höchsten Extinktionskoeffizienten aufwies, dem Uranyl(V) zugeordnet. Diese Wellenlänge wurde infolgedessen als Anregungswellenlänge für die TRLFS genutzt. Resultierend wurde ein Peakmaximum bei 440 nm der Uranyl(V)-Emission zugeordnet, mit einer Lebensdauer von 1.1 ± 0.021 µs.
Auch Grossmann et. al (2009) wiesen laut Literatur U(V) in der Form von [U(V)O2(CO3)3]5- nach.64 Damit der Komplex stabil vorliegt, wurde der pH-Wert auf 11.8 eingestellt. Die Anregungswellenlänge betrug hierbei 255 bzw. 408 nm. Bei einer Temperatur von 153 K wurden in der gefrorenen Lösung Uranyl(V)-Fluoreszenzbanden entdeckt, welche sich von 380-440 nm erstrecken, mit Maxima je nach Anregungswellenlänge um 404.7 und 413.3 nm.
Die Fluoreszenzlebensdauer bei 153 K betrug 120 μs. Ähnlich zu Uranyl(VI) wies der Uranyl(V)- Komplex hierbei eine Fluoreszenzbandenstruktur aus fünf Emissionsbanden auf, welche durch Spektrenentfaltung gewonnen wurden. Die beträchtliche Blauverschiebung, im Vergleich zu
26
den Ergebnissen von Steudtner et. al, wurde auf die komplexierenden Carbonatliganden zurückgeführt, welche auch in Uranyl(VI) hypsochromische Verschiebungen von etwa 15 nm verursachen.64
Arnold et. al (2010) untersuchten die Oxidation von UCl4 in Präsenz von Biofilmen und konnten mithilfe von Laserfluoreszenzmikroskopie eine Schrittweise Oxidation von U(IV) zu U(V) und letztendlich zu U(VI) beobachten.65 Für U(V) wurde eine Anregungswellenlänge von 408 nm genutzt, auf Basis von Grossmann et. al Ergebnissen. Das infolgedessen gemessene Fluoreszenzsignal im Bereich von 415-475 nm wurde als metastabiles U(V) auf der Grundlage von Steudtner et. al Resultaten identifiziert.
2.6.2. Uran(IV)
Das Uran(V), welches in 2.6.1 beschrieben wird, vollzieht eine Disproportionierung in Uranyl(VI) und Uran(IV). Entsprechend würde die Existenz von Uran(IV) mit hoher Wahrscheinlichkeit auf intermediär gebildetes Uranyl(V) hinweisen und die in 2.3 beschriebenen Theorien bestätigen.
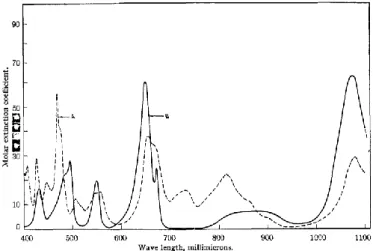
Abbildung 9: Von Kraus et. al gemessene Uran(IV)- (durchgezogene Linie) und Neptunium(IV)-UV/Vis-Spektren (gestrichelte Linie).66
Bei Uran(IV) handelt es sich um eine stabile Oxidationsstufe des Urans, welche hauptsächlich unter reduzierenden Bedingungen gebildet wird. Uran(IV) lässt sich als freies Aquo-Ion bis zu einem pH < 1.0 sehr gut mithilfe der UV/Vis-Absorptionsspektroskopie detektieren, was auf die höhere molare Absorptivität (ε(U(VI)) = 9.7 M-1 cm-1; ε(U(IV)) = 55 M-1 cm-1) von Uran(IV) zurückzuführen ist (siehe auch Abbildung 9).66-67 Ab einem pH von 1 setzt dann zunehmend
27
die Hydrolyse von Uran(IV) ein, was zu einer Änderung der UV/Vis-Absorptionsbanden führt.68-69
Mithilfe der TRLFS ist es möglich, das freie U(IV) zu detektieren, welches jedoch bei Raumtemperatur eine extrem kurze Lebensdauer von etwa 2.6 ns besitzt.70 Die Lebensdauer erhöht sich beträchtlich, wenn die Messung bei 77 K erfolgt.71 Kirishima et. al (2004) beobachteten im Emissionsspektrum des Uran(IV) zehn Banden: bei 525; 409; 394; 345; 338;
335; 320; 318; 291 und 289 nm.72 Während Komplexe von Uran(IV) mit Phosphat nachgewiesen werden konnten, war es noch nicht möglich, die erste Hydrolysespezies von Uran(IV) mithilfe der TRLFS zu beobachten.73
2.6.3. Radikalfänger
Einige der unter 2.3 aufgeführten Quenchingmechanismen von Uranyl(VI) mit Chlorid nehmen an, dass eine Rückoxidation von Uranyl(V) durch freie Biradikalanionen oder Hydroxyradikale erfolgt. Entsprechend würde eine Desaktivierung dieser Radikale eine Akkumulation von Uranyl(V) erlauben, was die Detektion von Uranyl(V) oder Uran(IV) erleichtern würde. Zur Desaktivierung von Radikalen werden Radikalfänger eingesetzt.
Mithilfe von Radikalfängern ist es möglich, freie chemische Radikale zu desaktivieren. Da Radikale die Eigenschaft haben ihr ungepaartes Elektron an ein anderes Molekül abzugeben, werden Radikalfänger zudem auch als Antioxidantien bezeichnet, da sie einen Oxidationsprozess des Radikals unterbinden.74 Dabei muss die Reaktion des Radikals mit dem Radikalfänger schneller ablaufen als die zu unterdrückende Nebenreaktion.75 Eine Art von Radikalfängern sind die stabilen Radikale, etwa das Stickstoffmonooxidradikal NO*, welches in Körpern von Lebewesen auftritt. Durch Radikalkombination des NO* mit einem anderen Radikal tritt eine Rekombination auf, wodurch eine Desaktivierung erfolgt.76
Eine andere Form von Radikalfängern sind Elemente oder Moleküle, welche Elektronen abgeben und damit sich selber oxidieren, um ein Radikal zu reduzieren. Diese Eigenschaft ist einigen d-Block Elementen zu eigen, zum Beispiel dem Mangan. Cheton et. al (1988) untersuchten das antioxidative Verhalten von Mangan(II) und ihre Resultate suggerierten, dass Mangan(II) eine inhibierende Wirkung auf Superoxidradikale O2-* hat.77 O2-* ähnelt
28
strukturell dem Dihalidradikalanion (siehe 2.3). Durch Reaktion von Mn2+ mit O2-* in der Gegenwart von Protonen erfolgt eine Oxidation des Mn(II) zu Mn(III) sowie die Bildung von Wasserstoffperoxid.
𝑂2−∗+ 𝑀𝑛2++ 2 𝐻+→ 𝐻2𝑂2+ 𝑀𝑛3+ (14)
Im weiteren Verlauf der Reaktion erfolgt eine erneute Bildung von Mn(II) durch die Oxidation des Wasserstoffperoxids zu Sauerstoff. In in-vitro Experimenten an Ratten konnten Hussain et. al (1999) aufzeigen, dass Mn bereits im nanomolaren Konzentrationsbereich Superoxidradikale und Hydroxyradikale HO* im mikromolaren Bereich desaktiviert.78 In diesem Fall wurde den Ratten MnCl2 verabreicht, welche in vitro als Mn-Superoxid-Dismutase vorliegt. Auch Coassin et. al (1992) waren in der Lage nachzuweisen, dass Mn(II) in der Lage ist, organische Carbonylradikale R-COO* zu reduzieren, indem Mn(II) zu Mn(III) oxidiert wird.75 Weiterhin ist auch Nickel als Radikalfänger von Interesse. In der Literatur wird Nickel nicht als klassischer Radikalfänger eingesetzt, verfügt jedoch über ein aktives Redoxverhalten, weshalb Nickel als Katalysator in organischen Kupplungsreaktionen eingesetzt wird.79-81 Nickel besitzt zudem die Eigenschaft, mit verschiedenen Hydroperoxiden unter Bildung von freien Radikalen zu reagieren. Burrows et. al zeigten zudem auf, dass Nickel, ähnlich dem Magnesium, kaum einen quenchenden Einfluss auf Uranyl(VI) hat.14 Aus diesem Grund ist es von Interesse, ob nicht auch Nickel als Radikalfänger in Kombination mit Uranyl(VI) fungiert.
Aromatische Moleküle mit funktionellen Hydroxygruppen eignen sich ähnlich zu den d-Elementen als Radikalfänger. Eines der bekanntesten ist das Hydrochinon, welches auch als Antioxidant bekannt ist. So wird Hydrochinon innerhalb der makromolekularen Chemie als Inhibitor genutzt, um den Beginn einer radikalischen Polymerisation zu verzögern oder ganz zu stoppen.82 Zudem konnte durch Kitagawa et. al (1992) gezeigt werden, dass Hydrochinon erfolgreich Superoxidradikale desaktiviert.83 Weiterhin eignet sich Hydrochinon auch als Radikalfänger für organische Moleküle, wie Thavasi et. al (2009) am Beispiel des DPPH- Radikals (DPPH = Diphenylpicrylhydrazyl) zeigen konnten.84




![Abbildung 18: UV/Vis-Absorptionsspektren von Uranyl(VI) bei pH = 0.5; I = 0.5 und [U] = 0.01 M und variierender Temperatur](https://thumb-eu.123doks.com/thumbv2/1library_info/4565389.1599815/46.892.163.722.513.968/abbildung-uv-vis-absorptionsspektren-uranyl-vi-variierender-temperatur.webp)
![Abbildung 19: a) UV/Vis-Absorptionsspektren von Uran(IV) bei pH = 0.5; I = 0.5 und [U] = 0.01 M und variierender Temperatur und b) vergrößerter Ausschnitt der Uranyl(VI)-Banden](https://thumb-eu.123doks.com/thumbv2/1library_info/4565389.1599815/47.892.242.645.362.1037/abbildung-absorptionsspektren-variierender-temperatur-vergrößerter-ausschnitt-uranyl-banden.webp)
