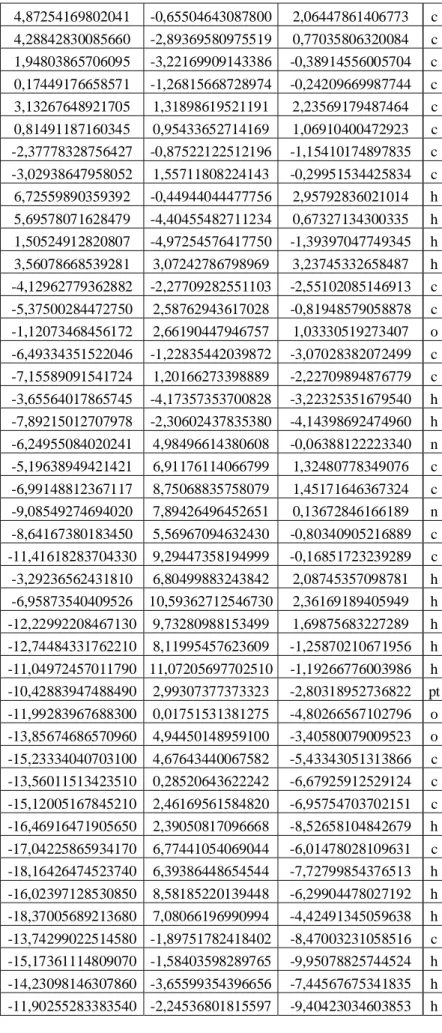
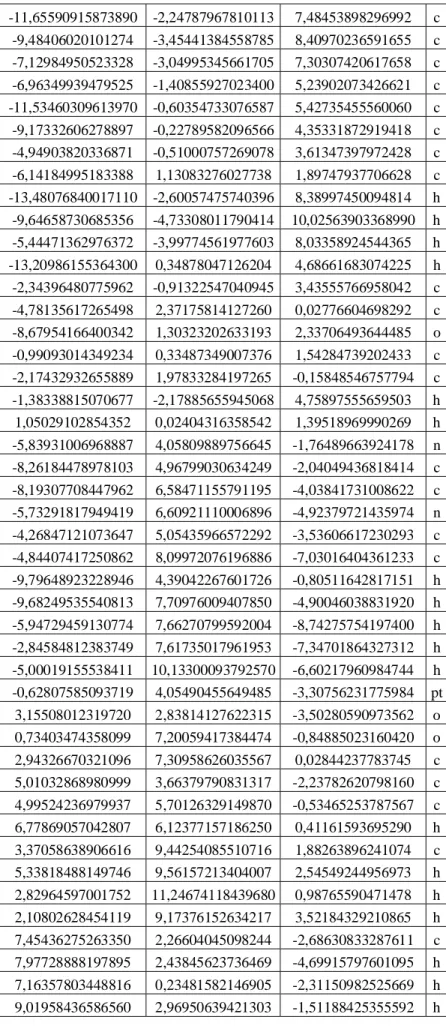
Emissionsverhaltens von unterschiedlich substituierten C^C* cyclometallierten Pt(II) Dibenzofuranyl-NHC-
Komplexen
Zur Erlangung des akademischen Grades Bachelor of Science (B. Sc.) im Studiengang Wirtschaftschemie
vorgelegt von Daniel Hansch
Düsseldorf, im Februar 2019
Durchgeführt am
Institut für Theoretische Chemie und Computerchemie Mathematisch-Naturwissenschaftliche Fakultät
Heinrich-Heine-Universität Düsseldorf
Erstgutachterin: Prof. Dr. Christel M. Marian Zweitgutachter: Prof. Dr. Christian Ganter
selbstständig ohne Hilfe Dritter verfasst und keine anderen als die angegebenen Quellen und Hilfsmittel benutzt habe. Die aus fremden Quellen direkt oder indirekt übernommenen Gedanken wurden als solche von mir kenntlich gemacht. Diese Arbeit hat in gleicher oder vergleichbarer Form noch keiner Prüfungsbehörde vorgelegen.
Düsseldorf, den 15.02.2019
_______________________________
Daniel Hansch
An erster Stelle möchte ich Frau Prof. Dr. Christel M. Marian für die Möglichkeit danken, diese Arbeit an einem interessanten Thema am Lehrstuhl für Theoretische Chemie und Computerchemie schreiben zu dürfen. Bedanken möchte ich mich auch für ihre Hilfestellungen und wertvollen Hinweise, die meine Arbeit vorangebracht haben.
Herrn Prof. Dr. Christian Ganter möchte ich für die Übernahme des Zweitgutachtens dieser Bachelorarbeit danken.
Ein weiterer Dank gilt meinen Betreuern Dr. Jelena Föller und Adrian Heil, die immer ansprechbar waren und mir bei diversen Fragen mit Rat und Tat hilfreich zur Seite standen.
Des Weiteren möchte ich mich beim ganzen Arbeitskreis der Theoretischen Chemie und Computerchemie für die angenehme Arbeitsatmosphäre und Unterstützung sowie für erheiternde Gespräche und Diskussionen auch abseits des Fachgebiets bedanken.
Ein besonderer Dank geht schließlich an meine Eltern, die mich während meines ganzen Studiums unterstützt und mir in schwierigen Situationen den Rücken gestärkt haben.
In dieser Arbeit werden zwei cyclometallierte Pt(II)-Komplexe, die sich nur in ihrer Substitution am Hilfsliganden unterscheiden und markant unterschiedliche Quantenausbeuten besitzen (90% und 0%), quantenchemisch untersucht und die Ergebnisse mit experimentellen Daten verglichen. Für beide Komplexe werden jeweils drei T1-Zustände mit unterschiedlichem Charakter optimiert und ihre energetische Lage auf TDDFT/TDA- und DFT/MRCI-Niveau dargestellt. Es stellt sich heraus, dass beide Komplexe einen MC-Zustand mit annähernd identischer Geometrie besitzen, bei der das Platinatom nicht mehr quadratisch-planar koordiniert ist und der Acetylacetonat- Hilfsligand sich aus der restlichen Ligandenebene herausdreht.
Des Weiteren werden für beide Komplexe Absorptionsspektren berechnet und mit den experimentellen Spektren verglichen. Hieraus geht hervor, dass sich unter Berücksichtigung der Spin-Bahn-Kopplung die berechneten Spektren den experimentellen Spektren annähern.
Für den emittierenden Komplex werden ausgehend vom energetisch niedrigsten T1- Zustand temperaturabhängige Emissionsspektren berechnet, wobei für eine Temperatur von 200 K das experimentelle Spektrum qualitativ gut wiedergegeben wird. Auch hier wird unter Einbezug der Spin-Bahn-Kopplung eine Verbesserung der Ergebnisse erzielt.
Es ist weiterhin gelungen, auch für den im Experiment nicht-emittierenden Komplex ausgehend von den beiden energetisch niedrigsten T1-Zuständen temperaturabhängige Emissionsspektren zu berechnen.
Weiterhin werden für beide Komplexe jeweils ausgehend von der Geometrie des energetisch niedrigsten T1-Zustandes Scans zur Geometrie des MC-Zustandes durchgeführt, um die Energiebarriere zwischen beiden Zuständen zu ermitteln.
Hierdurch soll das im Experiment unterschiedliche Emissionsverhalten beider Komplexe erklärt werden. Es stellt sich allerdings heraus, dass die Energiebarriere zum MC-Zustand, der für nicht strahlende Prozesse verantwortlich ist, beim nicht- emittierenden Komplex größer ist als beim emittierenden Komplex, was widersprüchlich ist.
Abkürzungsverzeichnis ... I
1. Einleitung ... 1
2. Theoretischer Hintergrund ... 2
2.1 Lumineszenz und photophysikalische Prozesse... 2
2.2 Aufbau und Funktionsweise einer OLED ... 3
2.3 Spinstatistik der Exzitonbildung ... 4
2.4 Grenzorbitale und Arten von Übergängen ... 5
3. Quantenchemische Methoden ... 6
3.1 Dichtefunktionaltheorie... 6
3.2 Verwendete Funktionale... 9
3.2.1 Das PBE0-Funktional ... 9
3.2.2 Das BH-LYP-Funktional ... 10
3.3 Zeitabhängige Dichtefunktionaltheorie/Tamm-Dancoff-Näherung ... 10
3.4 Multireference Configuration Interaction ... 11
3.5 DFT/MRCI ... 11
3.6 Effektives Kernpotential ... 13
3.7 Basissätze ... 13
4. Technische Details und Durchführung der Rechnungen ... 14
5. Ergebnisse und Auswertung ... 16
5.1 Komplex 1 ... 16
5.1.1 Die S0-Geometrie ... 16
5.1.2 Absorptionsspektren ... 18
5.1.3 Der S1-Zustand ... 21
5.1.4 Die T1-Zustände ... 22
5.1.4.1 Der T1a-Zustand ... 22
5.1.4.2 Der T1b-Zustand... 23
5.1.4.3 Der T1c-Zustand ... 23
5.1.5 DFT/MRCI-Rechnungen ... 25
5.1.6 Emissionsspektren ... 26
5.1.7 Scan von der T1a- zur T1c-Geometrie ... 28
5.2 Komplex 2 ... 30
5.2.1 S0-Geometrie ... 30
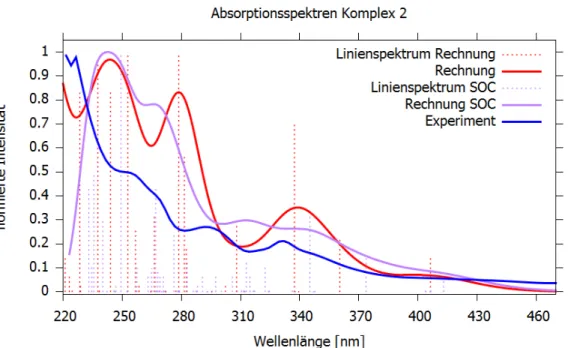
5.2.2 Absorptionsspektren ... 31
5.2.3 Der S1-Zustand ... 34
5.2.4 Die T1-Zustände ... 36
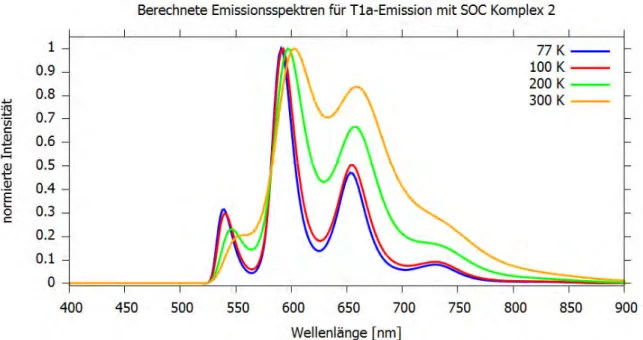
5.2.6 Emissionsspektren ... 40
5.2.7 Scan von der T1a- zur T1c-Geometrie ... 43
6. Diskussion und Fazit ... 44
Literaturverzeichnis ... i
Anhang ... iv
A. Tabellen ... iv
Komplex 1 ... iv
Komplex 2 ... xxxvii B. Abbildungen ... lxix Komplex 1 ... lxix Komplex 2 ... lxxxiii
Abkürzungsverzeichnis
Abb. Abbildung
acac Acetylacetonat
BH-LYP Becke Half and Half-Lee-Yang-Parr
bzw. beziehungsweise
CI Configuration Interaction
CSF Configuration State Functions
DBF Dibenzofuranyl
DFT Density Functional Theory
ECP Effective Core Potential
Eh Hartree-Energie
eV Elektronenvolt
fs Femtosekunden
GGA Generalized Gradient Approximation
HF Hartree-Fock
HOMO Highest Occupied Molecular Orbital
ILCT Intra-Ligand Charge Transfer
IR Infrared
Komplex 1 1-(Dibenzofuranyl)-3-methylimidazol-2-yliden- C2,C2´]platin(II)acetylacetonat
Komplex 2 1-(Dibenzofuranyl)-3-methylimidazol-2-yliden- C2,C2´]platin(II)hexafluoroacetylacetonat
KS Kohn-Sham
LC Ligand-centered
LCD Liquid Crystal Display
LDA Local Density Approximation
LLCT Ligand-to-Ligand Charge Transfer
LMCT Ligand-to-Metal Charge Transfer
LSDA Local Spin Density Aproximation
LUMO Lowest Unoccupied Molecular Orbital
MC Metal-centered
MLCT Metal-to-Ligand Charge Transfer
MRCI Multireference Configuration Interaction
OLED Organic Light Emitting Diode
PBE Perdew-Burke-Ernzerhof
PhOLED Phosphorescent Organic Light Emitting Diode
pm Picometer
PMMA Polymethylmethacrylat
Pt(II) Platin(II)
S0 Singulett-Grundzustand
S1 erster angeregter Sigulett-Zustand
SOC Spin-Orbit Coupling
SOCI Spin-Orbit Configuration Interaction
s. u. siehe unten
SV(P) Split-Valence (Polarization)
T1 erster angeregter Triplett-Zustand
TDA Tamm-Dancoff Approximation
TDDFT Time Dependent Density Functional Theory
UV/VIS Ultraviolet/Visible
1. Einleitung
Organische Leuchtdioden (OLEDs, engl.: Organic Light Emitting Diodes) haben in jüngster Vergangenheit sowohl bei der Fabrikation von neuartigen Displays in der Elektronikindustrie als auch in der Beleuchtungstechnik eine bedeutende Rolle eingenommen.[1] Nachdem Ching W. Tang und Steven Van Slyke im Jahre 1987 den Grundstein für die OLED-Technologie gelegt hatten,[2] wurden OLEDs im Laufe der Zeit kontinuierlich weiterentwickelt und hinsichtlich ihres technischen Aufbaus und des Materials, aus welchem sie bestehen, modifiziert. Der Markt für OLEDs hat in den letzten Jahren eine rasante Entwicklung erfahren und konkurriert mit LCDs (engl.:
Liquid Crystal Displays) in allen Anwendungen, vor allem im Bereich der kleinen Displays.[3] Ihr geringes Gewicht, eine dünnfilmige Konstruktion sowie flexible Materialeigenschaften machen OLEDs für Anwendungen in der Industrie interessant.[1]
Einen besonderen Fokus im Bereich der Industrie und der Forschung erfahren die sogenannten PhOLEDs (Phosphoreszierende organische Leuchtdioden). Diese beinhalten einen Übergangsmetallkomplex als Emitter und zeigen außergewöhnliche photophysikalische Eigenschaften. Als Übergangsmetallkomplexe dienen hierbei neutrale Iridium(III)- oder Platin(II)-Komplexe mit cyclometallierten Liganden, die zusätzlich ein erweitertes π-System am Ligandengerüst besitzen. Phosphoreszierende cyclometallierte Platin(II)-Komplexe weisen unter anderem hohe Quantenausbeuten auf, emittieren abhängig von der Art der Liganden bei verschiedenen Wellenlängen und zeigen unterschiedliche Emissionslebensdauern, was sie für Anwendungen in der Sensorik und in der biomedizinischen Technik nutzbar macht.[4]
In der Arbeitsgruppe Straßner (TU Dresden) wurden eine Reihe von cyclometallierten Pt(II)-Komplexen bestehend aus einem NHC-Dibenzofuranyl-Liganden sowie einem Acetylacetonat-Hilfsliganden synthetisiert. Es stellte sich heraus, dass chemisch ähnliche Komplexe in Abhängigkeit von den Substituenten am Hilfsliganden erstaunlicherweise markant unterschiedliche Quantenausbeuten aufzeigen.
Abb. 1: In der Arbeitsgruppe Straßner untersuchte cyclometallierte Platinkomplexe.
Links: Komplex 1: [1-(Dibenzofuranyl)-3-methylimidazol-2-yliden-C2,C2´]platin(II)acetylacetonat
Während Komplex 1 eine Quantenausbeute von 90% aufweist und im blau-grünen Bereich des sichtbaren Spektrums emittiert, ist Komplex 2 nicht emissiv.[5, 6] Für die beiden Komplexe wird vermutet, dass sie drei verschiedene Minima mit unterschiedlichem Charakter auf der T1-Potentialfläche besitzen.
Im Rahmen dieser Bachelorarbeit werden quantenchemische Rechnungen für beide Platin(II)-Komplexe durchgeführt, die Ergebnisse mit experimentellen Daten verglichen und Ursachen für das unterschiedliche photophysikalische Verhalten beider Komplexe untersucht.
2. Theoretischer Hintergrund
2.1 Lumineszenz und photophysikalische Prozesse
Lumineszenz beruht auf einem elektronischen Übergang von einem angeregten Zustand in den Grundzustand, wobei bei diesem Prozess elektromagnetische Strahlung im UV/VIS (engl.: Ultraviolet/Visible)- und IR (engl.: Infrared)-Bereich emittiert wird.[7]
Die Anregung der Elektronen kann auf unterschiedliche Arten erfolgen und führt zu verschiedenen Bezeichnungen der Lumineszenz.
Bei der Photolumineszenz beispielsweise erfolgt die Anregung durch Absorption von Photonen (Lichtabsorption). In OLEDs wird die Lichtemission durch das Anlegen einer elektrischen Spannung generiert. Diese Art der Anregung wird als Elektrolumineszenz bezeichnet. In allen Formen der Lumineszenz muss derselbe Betrag der Anregungsenergie ausgehend vom Grundzustand bei der Relaxation wieder an die Umgebung abgegeben werden. Die Rückkehr in den Grundzustand kann neben der Emission von Licht parallel auch durch Abgabe von Wärmeenergie erfolgen (s. u.).
Abb. 2: Jabłoński-Diagramm mit den möglichen Übergängen (schematisch).[8]
A = Absorption; F = Fluoreszenz; P = Phosphoreszenz;
IC = Interne Konversion; ISC = Intersystem Crossing;
VR = Schwingungsrelaxation
Anhand eines Jabłoński-Schemas (siehe Abb. 2) werden die zugrundeliegenden elektronischen Übergänge veranschaulicht.
Die meisten Moleküle besitzen im Grundzustand eine Singulett-Spinmultiplizität (S0).[8]
Kommt es nun zur Absorption von Energie, werden aus diesem Grund vorwiegend Singulettzustände vertikal angeregt (S1). Neben der elektronischen Anregung findet ebenfalls eine vibronische Anregung der Schwingungsniveaus des Moleküls statt. An die Anregung schließt sich die Relaxation in den Grundzustand an, wobei diese entweder durch Fluoreszenz (F) unter Abgabe von Photonen oder strahlungslos durch interne Konversion (IC) vollzogen wird. Die interne Konversion beschreibt einen Wechsel zwischen Zuständen gleicher Spinmultiplizität unter Energieerhalt. Auf die interne Konversion kann die vibronische Relaxation (VR) folgen, bei der die angeregten Schwingungsniveaus des Moleküls unter Abgabe von thermischer Energie in den Schwingungsgrundzustand des jeweiligen elektronischen Zustandes relaxieren.
Fluoreszenz, interne Konversion und vibronische Relaxation finden unter Beibehaltung der Spinmultiplizität statt und treten daher mit hoher Wahrscheinlichkeit auf.
Des Weiteren sind auch Übergänge möglich, bei denen sich die Spinmultiplizität ändert.
Zu dieser Kategorie zählt das Intersystem-Crossing (ISC). Hierbei schließt sich nach der Anregung des Singulettzustandes ein strahlungsloser Übergang in einen angeregten Triplettzustand (T1) an. Der angeregte Triplettzustand kann anschließend durch Phosphoreszenz (P) unter Abgabe von Photonen in den elektronischen Grundzustand (S0) zurückfallen. Intersystem-Crossing und Phosphoreszenz gehören zu den spin- verbotenen Übergängen und sind daher wenig wahrscheinlich. Über die Spin-Bahn- Kopplung, der Wechselwirkung zwischen Bahndrehimpuls und Elektronenspin, wird eine Lockerung des Spinverbots erreicht und Übergänge unter Änderung der Spinmultiplizität werden ermöglicht. Zu erwähnen ist, dass die Phosphoreszenz eine längere Lebensdauer der Lichtemission als die Fluoreszenz besitzt.
Durch die Spin-Bahn-Kopplung, welche bei Pt(II)-Komplexen eine Rolle spielt, werden die photophysikalischen Eigenschaften von PhOLEDs entscheidend beeinflusst (siehe Abschnitt 2.3).
2.2 Aufbau und Funktionsweise einer OLED
Eine OLED ist aus mehreren funktionalen Schichten zusammengesetzt. In Abb. 3 ist der schematische Aufbau einer OLED dargestellt, deren Funktionsweise im Folgenden erläutert wird.
Abb. 3: Schematischer Aufbau einer OLED [9].
Durch Anlegen einer elektrischen Spannung werden über die Anode Defektelektronen, sogenannte „Löcher“, in den Hole Injection Layer injiziert. Gleichzeitig gelangen die von der Kathode generierten Elektronen in den Electron Injection Layer. Die Löcher werden durch den Hole Transport Layer geleitet, während die Elektronen durch den Electron Transport Layer wandern. Der Hole Blocking Layer und der Electron Blocking Layer verhindern, dass Löcher bzw. Elektronen in die jeweiligen Transportschichten gelangen. Der Emissive Layer ist mit dem Emittermolekül dotiert. In dieser Schicht rekombinieren nun Löcher und Elektronen, es kommt zur Bildung von Exzitonen. Dabei werden die Emittermoleküle in einen angeregten Zustand versetzt und emittieren in der Folge Licht bei der Rückkehr in den Grundzustand.
2.3 Spinstatistik der Exzitonbildung
Löcher und Elektronen besitzen einen halbzahligen Spin ( . Die bei der Rekombination beider Ladungsträger entstehenden Exzitonen können somit einen Gesamtspin von 0 oder 1 besitzen. Ein Exziton mit 0 besitzt eine magnetische Spinquantenzahl von 0 und entspricht einem Singulett-Zustand. Der Gesamtspin mit 1 kann in drei Komponenten mit den magnetischen Spinquantenzahlen von 0, 1 und 1 aufgespalten werden, aus denen Triplett- Zustände resultieren. Für die Bildung eines Exzitons sind somit vier Spinkombinationen
möglich, wobei jede Kombination mit der gleichen Wahrscheinlichkeit gebildet werden kann. Hieraus ergibt sich die Spinstatistik, nach welcher 25 % Singulett-Exzitonen und 75 % Triplett-Exzitonen geerntet werden können. Über die Spin-Bahn-Kopplung ist es somit möglich, eine theoretische Quantenausbeute von 100 % zu erreichen.[6]
2.4 Grenzorbitale und Arten von Übergängen
Ein einfaches Grenzorbital-Modell bietet einen Überblick über die möglichen emittierenden Zustände von Übergangsmetallkomplexen. Für die Betrachtung ist es zweckmäßig, sich auf die an den Liganden lokalisierten besetzten π- und unbesetzten π*-Orbitale sowie auf die besetzten d- und unbesetzten d*-Orbitale am Metallzentrum zu beschränken. In Abhängigkeit von der Art der Anregung aus dem elektronischen Grundzustand lassen sich verschiedene Übergänge charakterisieren, die im Folgenden näher beschrieben werden.
Ein ligandenzentrierter Zustand, der als LC (ππ*) (Ligand-centered) abgekürzt wird, entsteht durch die Anregung eines Elektrons aus einem π- in ein π*-Orbital. Wird während des Übergangs Ladungsdichte innerhalb eines Liganden verschoben, spricht man von einem ILCT-Übergang (Intra-Ligand Charge Transfer). Findet der Übergang zwischen π- und π*-Orbitalen verschiedener Liganden statt, so wird der Vorgang als LLCT-Übergang (Ligand-to-Ligand Charge Transfer) charakterisiert.
Beim metallzentrierten Übergang kommt es zur Anregung aus einem d- in ein d*- Orbital, abgekürzt als MC (dd*) (Metal-centered). Hier sollte erwähnt werden, dass metallzentrierte Triplett-Zustände bei Pt(II)-Komplexen typischerweise für nichtstrahlende Desaktivierungsprozesse verantwortlich sind.[4] Schließlich sind noch Übergänge möglich, bei denen entweder ein elektronischer Übergang vom Zentralmetallatom zum Liganden (dπ*) oder vom Liganden zum Zentralmetallatom (πd*) stattfindet. In diesen Fällen spricht man von MLCT (dπ*) (Metal-to-Ligand Charge Transfer)- bzw. LMCT (πd*) (Ligand-to-Metal Charge Transfer)-Übergängen.
Unter Berücksichtigung von Wechselwirkungen zwischen den Elektronen des jeweiligen Zustands findet eine Aufhebung der energetischen Entartung der Zustände statt.[10] Die Triplett-Zustände werden hierbei aufgrund der Austauschwechselwirkung der Elektronen energetisch gegenüber dem Singulett-Zustand abgesenkt.
3. Quantenchemische Methoden
3.1 Dichtefunktionaltheorie
Die Dichtefunktionaltheorie (DFT) besagt, dass die exakte Energie eines molekularen Systems im Grundzustand eine Funktion der Elektronendichte und der Positionen der Atomkerne ist. Mittels Integration der Elektronendichte über den gesamten Raum ergibt sich die Anzahl der Elektronen.[11]
(1)
Nach dem 1. Theorem von Hohenberg und Kohn gilt, dass die elektronische Grundzustandsenergie eindeutig durch die Einelektronendichte beschrieben werden kann.[12] Das Funktional , welches die Einelektronendichte mit der elektronischen Grundzustandsenergie verknüpft, wird als Dichtefunktional bezeichnet. Das 2. Theorem von Hohenberg und Kohn besagt, dass für dieses Dichtefunktional ein Variationsprinzip gilt.[12] Die berechnete Energie ist immer größer oder gleich der wahren Grundzustandsenergie des Systems. Das exakte Funktional, das die Dichte mit der Energie verknüpft, ist allerdings nicht bekannt.
Die Dichtefunktionaltheorie bietet den Vorteil, dass die Elektronendichte unabhängig von der Anzahl der Elektronen ist. Im Gegensatz zur elektronischen Wellenfunktion, die mit 4 Variablen (Elektronenspin und die drei Raumrichtungen) arbeitet, handelt es sich bei der gesuchten Dichte um eine Funktion von drei Ortskoordinaten.
Da es keine exakte analytische Vorschrift zur direkten Berechnung der Energie aus der Einelektronendichte gibt, wurden Ansätze entwickelt, um zu einer genäherten Lösung zu gelangen. Das Dichtefunktional lässt sich analog zur wellenfunktionsbasierten Schrödinger-Gleichung für ein Vielteilchensystem in Komponenten aufteilen:
(2)
ist das Funktional der kinetischen Energie, ein Funktional für die Coulomb-Wechselwirkung zwischen Atomkernen und Elektronen, dient als Funktional zur Beschreibung der Coulomb-Abstoßung der Elektronen untereinander, während das Funktional die Austauschwechselwirkung repräsentiert. und
lassen sich mittels klassischer Physik beschreiben:
| | (3)
1
2 | | (4)
ist die Kernladungszahl, steht für die Kernkoordinate und bzw. sind die Ortsvariablen des Elektrons.
Für die kinetische Energie und die Austauschwechselwirkung können keine expliziten Terme angegeben werden. W. Kohn und L. J. Sham haben im Jahre 1965 einen Ansatz entwickelt mit dem Ziel, die kinetische Energie so gut wie möglich zu approximieren.[13] Im sogenannten Kohn-Sham-Formalismus wird hierzu das Funktional der kinetischen Energie in einen nichtwechselwirkenden Term und einen Korrelationsterm aufgespalten.
(5)
Für gilt die Annahme eines nichtwechselwirkenden Systems von Elektronen.
Hierfür werden als Hilfsgrößen Einteilchenorbitale , die Kohn-Sham-Orbitale, eingeführt. Über Quadrierung und Aufsummierung der Orbitale ergibt sich folgender approximativer Ausdruck für die Dichte von Elektronen:
| | (6)
Die kinetische Energie wird dann folgendermaßen beschrieben:
1
2 | | (7)
Für die Beschreibung der Wechselwirkung zwischen den Teilchen wird das Austauschkorrelationsfunktional eingeführt, das Korrekturen für die Zwei- Elektronen-Austauschwechselwirkung, die dynamische Elektronenkorrelation und die kinetische Energie enthält. Es setzt sich folgendermaßen zusammen:
(8)
Das Kohn-Sham-Dichtefunktional ergibt sich damit zu
= + (9)
Die Berechnung der Kohn-Sham-Orbitale zur Ermittlung der Elektronendichte erfolgt durch Lösen der Eigenwertgleichung
(10) wobei
1
2 (11)
gilt. Der Kohn-Sham-Operator enthält das Kohn-Sham-Potential , welches die Summe aus dem attraktiven Kern-Elektron-Potential, dem klassischen Coulomb- Wechselwirkungspotential und dem Austauschwechselwirkungspotential bildet:
| | (12)
Die Gleichungen (10), (11) und (12) stellen die Kohn-Sham-Gleichungen dar, deren Lösung iterativ erfolgt. Man geht hierbei zunächst von einer Ausgangsdichte
aus, berechnet und löst anschließend die Eigenwertgleichung (10) für die Orbitale . Ausgehend von diesem Ergebnis kann wieder ein neues Potential
berechnet werden und Gleichung (10) erneut gelöst werden. Das Verfahren wird bis zur Selbstkonsistenz wiederholt. Die Funktionen zu einem Satz niedrigster Orbitalenergien stellen die gesuchten Kohn-Sham-Orbitale dar.[11]
Für die Beschreibung des Austauschkorrelationsfunktionals gibt es verschiedene Näherungsverfahren. Die lokale Dichtenäherung (LDA, engl.: Local Density Approximation) beinhaltet eine Näherung des Austauschkorrelationsfunktionals über die Behandlung der Elektronendichte als uniformes Elektronengas. Die Austauschkorrelationsenergie ist in dem Modell an jedem Ort des Systems die Gleiche, da das Elektronengas überall dieselbe Dichte besitzt. Dieser Ansatz geht von einem nicht-wechselwirkenden Elektronengas aus. Bei der lokalen spinabhängigen Dichtenäherung (LSDA, engl.: Local Spin Density Approximation) wird zusätzlich der Elektronenspin berücksichtigt. Die verallgemeinerte Gradientennäherung (GGA, engl.:
Generalized Gradient Approximation) berücksichtigt nicht nur die Funktion der Elektronendichte an einem bestimmten Ort, sondern auch ihre Ableitungen nach dem Ort. Sogenannte Hybridfunktionale (siehe Abschnitt 3.2) kombinieren das Austauschkorrelationsfunktional mit exaktem Hartree-Fock-Austausch.
3.2 Verwendete Funktionale
3.2.1 Das PBE0-Funktional
Im PBE0-Funktional wird das PBE (Perdew-Burke-Ernzerhof)-Funktional, welches auf der GGA basiert, in Kombination mit einem exakten Austauschterm verwendet.[14]
1
4 (13)
Über die Terme und werden die Austausch-Korrelations- und Austausch- Beiträge aus der GGA beschrieben.
3.2.2 Das BH-LYP-Funktional
Das BH-LYP (Becke Half and Half-Lee-Yang-Parr)-Funktional beschreibt die Austauschkorrelationsenergie zu 50 % durch einen Hartree-Fock-Austauschterm und zu 50 % durch ein Becke-Funktional (B88) in Kombination mit einem LYP- Korrelationsterm.[15]
0,5 0,5 (14)
3.3 Zeitabhängige Dichtefunktionaltheorie/Tamm-Dancoff-Näherung
Die zeitabhängige Dichtefunktionaltheorie (TDDFT, engl.: Time Dependent Density Functional Theory) ist eine Erweiterung des DFT-Formalismus und wird häufig für die Berechnung von angeregten Zuständen genutzt. In dieser Theorie wird zusätzlich der Einfluss eines externen Potenzials auf die zeitabhängige Veränderung der Elektronendichte betrachtet. Die TDDFT ermöglicht also einen Zugang zur Beschreibung von dynamischen Phänomenen. Sie kann somit konkret angewendet werden, um den Einfluss eines elektrischen Feldes auf die Anregung von OLED- Emittern zu beschreiben.
Grundlage dieser Theorie ist das sogenannte Runge-Gross-Theorem, welches im Jahre 1984 aufgestellt wurde[16] und eine zeitabhängige Erweiterung des ersten Hohenberg- Kohn-Theorems aus der DFT darstellt. Das Runge-Gross-Theorem besagt, dass das zeitabhängige externe Potential eines Systems eindeutig seiner zeitabhängigen Dichte zugeordnet werden kann. Analog zur DFT erfolgt auch hier die Einführung eines Kohn- Sham-Referenzsystems nicht-wechselwirkender Elektronen. Die Kohn-Sham- Gleichungen resultieren aus der zeitabhängigen Schrödinger-Gleichung.
Die Mängel in der TDDFT liegen darin begründet, dass Anregungsenergien für Charge-Transfer-Übergänge aufgrund der falschen asymptotischen Beschreibung der langreichweitigen Wechselwirkung systematisch unterschätzt werden.[17]
Die Tamm-Dancoff-Näherung (TDA, engl.: Tamm-Dancoff Approximation) ist ein Näherungsverfahren zur TDDFT und stellt eine gute Alternative zur Behandlung großer Moleküle dar, da die TDDFT bei der Berechnung jener zeit- und kostenintensiv ist. Die Anwendung der TDA führt zu einer erheblichen Reduktion des Rechenaufwandes.[18]
3.4 Multireference Configuration Interaction
Der CI (engl.: Configuration Interaction)-Ansatz stellt eine Methode zur Lösung der Schrödinger-Gleichung dar. Hierbei wird die Vielteilchen-Wellenfunktion aus einer Linearkombination von Slater-Determinanten, den CSFs (engl.: Configuration State Functions) entwickelt.[19] Dies ermöglicht es, sowohl die Energie des Grundzustandes als auch der angeregten Zustände unter Berücksichtigung unterschiedlicher Elektronenkonfigurationen zu berechnen.
⋯ (15)
Hier wird von einer Referenzwellenfunktion ausgegangen. Durch den zweiten Term wird eine Einfachanregung aus einem besetzten Orbital in ein unbesetztes virtuelles Orbital beschrieben. Der dritte Term steht dann entsprechend für eine Doppelanregung.
Die Terme werden mit dem Koeffizienten variationell gewichtet. Aus Gleichung (15) lässt sich eine CI-Matrix bilden, aus der die Eigenwerte berechnet werden können.
Gleichung (15) kann je nach Art der Anregung um weitere Summanden ergänzt werden.
In dem MRCI (engl.: Multireference Configuration Interaction)-Ansatz werden Mehrfachanregungen ausgehend von mehreren Referenzdeterminanten verschiedener Konfigurationen durchgeführt. MRCI eignet sich deswegen gut zur Erfassung der statischen Elektronenkorrelation.
In einem Full-CI können alle möglichen Konfigurationen der Anregungen beschrieben werden. Bei einer vollständigen Basis würde dies zu einer exakten Bestimmung der Energie eines Systems führen. Da der Rechenaufwand vor allem bei großen Molekülen unverhältnismäßig hoch werden kann, werden die Anregungen auf einen sogenannten aktiven Raum beschränkt. In der Praxis werden hierfür kernnahe und energetisch hoch liegende Orbitale für die Rechnungen „eingefroren“, da diese für Anregungen kaum eine Rolle spielen.
3.5 DFT/MRCI
Die Kombination aus DFT und MRCI ermöglicht die Beschreibung der dynamischen Elektronenkorrelation aus der DFT sowie der statischen Elektronenkorrelation aus der
MRCI. Die aus Gleichung (15) resultierende CI-Matrix kann in Diagonal- und Außerdiagonalelemente aufgeteilt werden. Da die Diagonalelemente den größten Energiebeitrag liefern, ist die CI-Rechnung „diagonal-dominant“. Die Diagonalelemente bestehen aus Hartree-Fock (HF)-Ausdrücken und einem DFT- spezifischen Korrekturterm. Coulomb- und Austauschintegrale werden jeweils durch einen Parameter angepasst. Es ergibt sich folgender Ausdruck:[15]
∈
∈
(16)
bezeichnet die Anzahl der Anregungen, während die Indizes a und c sich auf vernichtete und erzeugte Elektronen beziehen. Da Teile der exakten Matrix-Elemente als Summe über alle Orbitalenergien ausgedrückt werden können, werden in den ersten beiden Summanden der Gleichung (16) die HF-Orbitalenergien durch ihre Kohn-Sham (KS)-Gegenstücke ersetzt.
Um eine Doppeltzählung der dynamischen Korrelation zu vermeiden, muss der Hamilton-Operator entsprechend parametrisiert werden. Hierfür werden die Coulomb- und Austauschterme der Diagonalelemente (16) modifiziert und die Außerdiagonalelemente in Abhängigkeit von ihrer Energiedifferenz mittels einer Dämpfungsfunktion skaliert.[15] Dabei wird die Wechselwirkung energetisch hochliegender Konfigurationen gedämpft, was zur Folge hat, dass viele Wechselwirkungen bereits im Vorfeld aussortiert werden können. Somit wird der Konfigurationsraum verkleinert und die Rechnungen laufen schneller ab.
In dieser Bachelorarbeit wurde für die DFT/MRCI-Rechnungen ein in der Arbeitsgruppe Marian neu parametrisierter Hamilton-Operator, der R2018, genutzt. Er besitzt folgende Dämpfungsfunktion:[20]
∙exp ∙ ∙ (17)
wobei den Skalierungsfaktor darstellt und für die Energiedifferenz zwischen den Außerdiagonalelementen steht.
Der R2018 liefert eine verbesserte Beschreibung von Übergangsmetallkomplexen. Der mittlere Fehler bei der Berechnung der relativen Energien beläuft sich hier auf 0,15 eV.[20] Es hat sich zudem herausgestellt, dass die Anregungsenergien von Übergangsmetallverbindungen durch die Dämpfungsfunktion deutlich beeinflusst werden.[20]
3.6 Effektives Kernpotential
Da vor allem bei Elementen höherer Perioden die Rumpfelektronen kaum Einfluss auf die chemische Bindung in einem Molekül ausüben, hat es sich als zweckmäßig erwiesen, diese in einer Näherung durch ein Pseudopotential zu beschreiben, um den Rechenaufwand zu verringern. Dieses Pseudopotential fließt als Einelektronenanteil in den Hamilton-Operator mit ein.[11] Das ECP (engl.: Effective Core Potential) beschreibt in einer Näherung die Wechselwirkung zwischen den Rumpfelektronen untereinander sowie die Wechselwirkung zwischen Rumpf- und Valenzelektronen. Das Pseudopotential enthält zudem skalarrelativistische Effekte, da kernnahe Elektronen eine Veränderung ihrer Masse erfahren. So entspricht die Geschwindigkeit eines Elektrons in Kernnähe für schwere Elemente nahezu der Lichtgeschwindigkeit, wodurch die Masse der Elektronen zunimmt. Daraus resultiert eine stärkere Anziehung zwischen Kern und Elektron, was wiederum zu einer Kontraktion des beteiligten Orbitals führt. In der Folge werden äußere Elektronen stärker vom Kern abgeschirmt.
Kernnahe Orbitale werden energetisch abgesenkt, während äußere Orbitale energetisch angehoben werden. In dieser Bachelorarbeit wurde für das Platin-Atom das skalarrelativistische effektive Rumpfpotential def-ECP verwendet.[21]
Hierbei werden die ersten 60 Elektronen (1s bis 4f) des Platins durch dieses Pseudopotential beschrieben.
3.7 Basissätze
Ein Basissatz ist ein Satz mathematischer Funktionen, aus denen die Wellenfunktion konstruiert wird. Jedes Molekülorbital wird als Linearkombination der Basisfunktionen an den atomaren Zentren dargestellt. Die Basisfunktionen selbst stellen Linearkombinationen von Gaußfunktionen dar. Die individuellen Funktionen werden als primitive Gaußfunktionen bezeichnet. Primitive Gaußfunktionen können über
Linearkombination zu kontrahierten Gaußfunktionen zusammengefasst werden, was die Beschreibung der kernnahen Bereiche eines Atoms verbessert.
Beim def-SV(P)-Basissatz (Split Valence mit Polarisation),[22, 23] der in dieser Arbeit verwendet wurde, werden die Rumpf-Atomorbitale durch eine kontrahierte Gaußfunktion beschrieben, während die Valenz-Atomorbitale durch zwei kontrahierte Gaußfunktionen dargestellt werden. Zusätzlich wird für alle Nicht-Wasserstoffatome ein Satz von Polarisationsfunktionen verwendet. Für die Atome der in dieser Arbeit berechneten Komplexe ergeben sich folgende Kontraktionsschemata:
Tabelle 1: Kontraktionsschemata der Atome der untersuchten Komplexe.
Atom Kontraktionsschema C (7s, 4p, 1d) -> [3s, 2p, 1d]
H (4s) -> [2s]
O (7s, 4p, 1d) -> [3s, 2p, 1d]
N (7s, 4p, 1d) -> [3s, 2p, 1d]
Pt (7s, 6p, 5d) -> [6s, 3p, 2d]
F (7s, 4p, 1d) -> [3s, 2p, 1d]
4. Technische Details und Durchführung der Rechnungen
Zur Konstruktion der beiden Komplexe wurde das Programm Avogadro 1.1.1[24, 25]
genutzt. Die Geometrieoptimierungen der Komplexe wurden mit Turbomole 7.0[26]
durchgeführt. Hierbei wurden das PBE0-Dichtefunktional (siehe Abschnitt 3.2.1), eine def-SV(P)-Basis für die Liganden (siehe Abschnitt 3.7) und das skalarrelativistische effektive Rumpfpotenzial def-ECP (siehe Abschnitt 3.6) mit dem zugehörigen def- SV(P)-Basissatz für das Platinatom verwendet (siehe Abschnitt 3.7). Für die Grundzustände erfolgte die Optimierung mit DFT unter Verwendung des jobex- Skriptes[27]. Die Schwingungsfrequenzanalysen zur Verifizierung, ob es sich bei den optimierten Strukturen um Minima handelt, wurden mit SNF V 5.0.1[28] erstellt. Für die Optimierung des ersten angeregten Singulettzustands und der ersten angeregten Triplettzustände wurde TDDFT bzw. TDDFT/TDA benutzt.[29–32] Mittels DFT/MRCI[15,
20, 33]
(siehe Abschnitt 3.5), das für das BH-LYP-Funktional (siehe Abschnitt 3.2.2) parametrisiert ist, wurden an den optimierten Strukturen des S0-Grundzustandes beider
Komplexe 41 Singulett- und 40 Triplettzustände berechnet. An den optimierten Strukturen der ersten angeregten Singulett- und Triplettzustände erfolgten mit dem DFT/MRCI-Programm jeweils Berechnungen von 21 Singulett- und 20 Triplettzuständen. Für alle DFT/MRCI-Rechnungen wurde der in der Arbeitsgruppe Marian neu parametrisierte Hamiltonian R2018 verwendet (siehe Abschnitt 3.5). Zudem wurden für diese Rechnungen mittels des Befehls rimp2prep jeweils alle Orbitale mit einer Energie größer als +2 Eh sowie alle Rumpforbitale mit einer Energie kleiner als -3 Eh eingefroren. Der erste Durchlauf der DFT/MRCI-Rechnung, der dem Auffinden der Referenzkonfigurationen dient, erfolgte jeweils mit einem Selektionsschwellenwert von 0,8 Eh. Im Input wurde hierbei festgesetzt, dass Einfach- und Doppelanregungen von zehn Elektronen in zehn Grenzorbitalen berechnet werden sollen. Für den zweiten Durchlauf wurde der Selektionsschwellenwert auf 1,0 Eh gesetzt. Alle DFT/MRCI- Rechnungen erfolgten ausgehend von einer C1-Symmetrie der Komplexe. Die Berechnung der Zustände wurde also jeweils für eine irreduzible Darstellung durchgeführt. Für die Einzelpunktrechnungen wurde dscf [34] verwendet.
Mit dem Programm plotter wurde eine Gaußverbreiterung der über DFT/MRCI berechneten Linienspektren erstellt. Über gnuplot 5.2.0[35] wurden die Absorptions- und Emissionsspektren geplottet. Die Digitalisierung der experimentellen Absorptionsspektren erfolgte mit dem Programm Engauge Digitizer 10.11.[36] Mit jmol 14.4.4[37] wurden die Geometrien der Komplexe sowie die Orbital- und Differenzdichten visualisiert. Für die Visualisierung der Orbitale bzw. Differenzdichten wurde hierbei ein cutoff von 0,03 bzw. 0,001 verwendet. Eine manuelle Atomnummerierung sowie die farbliche Hervorhebung von Bindungen und Bindungswinkeln erfolgte mit dem Bildbearbeitungsprogramm gimp 2.10.8.[38]
Für beide Komplexe wurden mit VIBES 1.2[39] temperaturabhängige Franck-Condon- Emissionsspektren berechnet. Dabei wurden für den Input folgende Einstellungen vorgenommen: Anzahl der Gitterpunkte: 16384; Intervall: 300 fs; Dämpfung: 75 cm-1. Als Temperaturen wurden analog zu den experimentell aufgenommenen Emissionsspektren 77 K, 100 K, 200 K und 300 K gewählt.
Die Koordinaten für den Scan der T1c-Geometrie (siehe Abschnitt 5.1.7 und 5.2.7) wurden mit der graphischen Benutzeroberfläche TmoleX16 erzeugt.
5. Ergebnisse und Auswertung
Komplex 1 und Komplex 2 wurden von der Arbeitsgruppe Straßner (TU Dresden) synthetisiert.[5, 6] Bindungslängen und –winkel dieser Komplexe wurden mittels einer Kristallstrukturanalyse bestimmt.[5,6]
Ebenso wurden Absorptions- und Emissionsspektren für beide Komplexe aufgenommen.[5, 6] In dieser Bachelorarbeit erfolgt ein Vergleich der durchgeführten quantenchemischen Rechnungen an beiden Komplexen mit den experimentellen Daten.
Aus den Ergebnissen der Rechnungen soll eine Erklärung für das unterschiedliche photophysikalische Verhalten (siehe Abschnitt 1) beider Komplexe gefolgert werden.
5.1 Komplex 1
5.1.1 Die S0-Geometrie
In Tabelle 2 und 3 werden die experimentell ermittelten Geometrieparameter[5] mit den berechneten Bindungslängen und –winkeln des mit DFT optimierten S0-Grundzustandes von Komplex 1 verglichen.
Abb. 4.: Struktur (nummeriert) der berechneten S0-Geometrie von Komplex 1. Die Farben stehen für folgende Atome: grau: Kohlenstoff; rot: Sauerstoff; blau: Stickstoff; weiß: Wasserstoff; hellgrau: Platin
Tabelle 2: Vergleich von berechneten und experimentell ermittelten Bindungslängen.
betrachtete Bindung
berechnete Bindungslängen [pm]
experimentell ermittelte Bindungslängen (gerundet) [pm][5]
Differenz Δ [pm]
Pt-C2 196 194 +2
Pt-C1 199 196 +3
Pt-O1 208 206 +2
Pt-O2 214 209 +5
Tabelle 3: Vergleich von berechneten und experimentell ermittelten Winkeln.
betrachtete Bindung berechnete Bindungswinkel [°]
experimentell ermittelte
Bindungswinkel [°][5] Differenz Δ [°]
C2-Pt-C1 80,2 80,5 -0,3
O2-Pt-O1 87,9 90,0 -2,1
C1-Pt-O1 92,9 91,4 +1,5
C2-Pt-O2 99,0 98,0 +1,0
C2-N2-C11-C1 -0,1 -2,8 +2,7
Sowohl in der Rechnung als auch in der Kristallstrukturanalyse liegt Komplex 1 annähernd planar (siehe Tabelle 3, Diederwinkel C2-N2-C11-C1) vor. Zwischen den experimentell ermittelten und den berechneten Werten bestehen geringfügige Abweichungen. Bei Betrachtung der Bindungslängen fällt auf, dass die Pt-O-Bindungen sowohl in der Rechnung als auch im Experiment im Allgemeinen größer als die Pt-C- Bindungen sind. Die berechneten Bindungslängen für die vorliegenden Bindungen sind zudem generell größer als die experimentell ermittelten Bindungslängen (siehe Tabelle 2).
Bei den Bindungswinkeln ergibt sich die Auffälligkeit, dass sowohl in der Rechnung als auch im Experiment die Winkel zwischen dem Platin und den beiden Sauerstoff- Atomen deutlich größer sind als die Winkel zwischen Platin und den beiden Kohlenstoff-Atomen (siehe Tabelle 3). In der experimentell ermittelten Kristallstruktur liegt der Winkel zwischen Platin und den Sauerstoff-Atomen bei 90°, was ideal für eine quadratisch-planare Koordination des Platins ist. In der berechneten S0-Geometrie ist dieser Winkel hingegen um 2,1° kleiner.
5.1.2 Absorptionsspektren
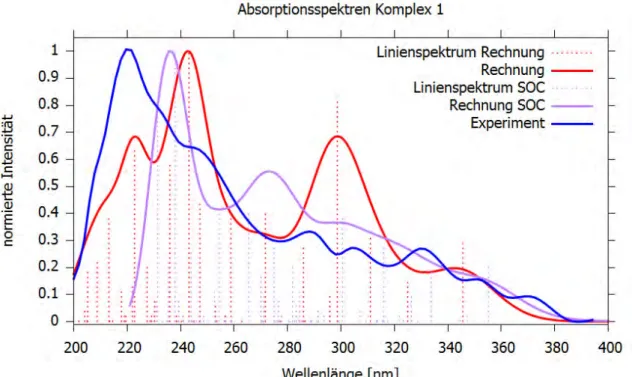
Ausgehend von DFT/MRCI-Rechnungen am S0-Grundzustand wurden Absorptionsspektren mit und ohne Spin-Bahn-Kopplung für Komplex 1 berechnet.
Diese sind in Abbildung 5 gemeinsam mit dem experimentellen Absorptionsspektrum dargestellt. Mittels des DFT/MRCI-Programms wurden zum einen ohne Einbezug der Spin-Bahn-Kopplung 41 Singulettzustände berechnet, um den Wellenlängenbereich des experimentellen Absorptionsspektrums zu plotten. Die Berechnung des Absorptionsspektrums unter Einfluss der Spin-Bahn-Kopplung (SOC, engl.: Spin-orbit coupling) wurde von Frau Prof. Marian durchgeführt. Dies erfolgte störungstheoretisch auf der Basis von 21 Singulett- sowie 20 Triplett-Zuständen, wobei die Triplett- Zustände in jeweils drei Zustände aufgespalten werden. Für die umhüllende Kurve des berechneten Linienspektrums wurde jeweils eine Halbwertsbreite von 1000 cm-1 gewählt.
Das experimentelle Absorptionsspektrum wurde bei einer Komplex-Konzentration von 2 Massen-% in einem PMMA (Polymethylmethacrylat)-Film bei Raumtemperatur aufgenommen.[5]
Um Vergleichbarkeit zu gewährleisten, sind alle Spektren auf eine Intensität von 1 normiert. Der dargestellte Wellenlängenbereich entspricht dem des experimentellen Spektrums.
Abb. 5: Berechnetes Absorptionsspektrum (rot) und berechnetes Absorptionsspektrum mit Spin-Bahn- Kopplung (violett) mit einer Halbwertsbreite von jeweils 1000 cm-1 sowie das experimentelle
Im berechneten Absorptionsspektrum ohne eingeschaltete Spin-Bahn-Kopplung können drei Peaks identifiziert werden, für die sich helle Übergänge mit den größten Oszillatorstärken im Spektrum verantwortlich zeigen (siehe Abb. 5). Da die Anregungen häufig einen gemischten Charakter aufweisen, empfiehlt es sich, die Übergänge durch Differenzdichten zu veranschaulichen. Die roten Flächen entsprechen hierbei einer Abnahme von Elektronendichte, während die blauen Flächen eine Zunahme von Elektronendichte bei der Anregung anzeigen. Die Differenzdichten für die drei hellsten Übergänge sind in Abb. 6 dargestellt.
Der erste Peak bei einer Wellenlänge von etwa 223 nm mit einer relativen Intensität von 68% entspricht einer Anregung von S0 nach S25 mit einer Oszillatorstärke von 0,30311.
Die Elektronendichte verlagert sich hierbei größtenteils auf den äußeren Phenylring des DBF-Liganden. Außerdem kommt es zu einer Abnahme der Elektronendichte am Platinatom (siehe Abb. 6). Die Anregung weist somit einen gemischten LC/MLCT- Charakter auf. Für die an der Anregung beteiligten Orbitale besitzt der Übergang von HOMO-2 (HOMO, engl.: Highest Occupied Molecular Orbital) nach LUMO+2 (LUMO, engl.: Lowest Unoccupied Molecular Orbital) mit 26,62% den größten Anteil an der Gesamtwellenfunktion.
Der zweite und größte Peak im berechneten Absorptionsspektrum ohne Spin-Bahn- Kopplung kann bei einer Wellenlänge von 243 nm ausfindig gemacht werden und entspricht einer Anregung nach S17 mit einer Oszillatorstärke von 0,47149. Hier verlagert sich die Elektronendichte zum größten Teil auf den NHC-Liganden, wobei es gleichzeitig zu einer Abnahme an Elektronendichte am Platin und an den restlichen Liganden kommt (siehe Abb. 6). Die Anregung besitzt einen LLCT/MLCT-Charakter.
Der wichtigste Übergang ist hier eine Anregung von HOMO nach LUMO+3 mit einem Anteil von 31,30% an der Gesamtwellenfunktion.
Der dritte und letzte Peak bei einer Wellenlänge von 299 nm mit einer relativen Intensität von 68% steht für eine Anregung nach S4 mit einer Oszillatorstärke von 0,38622. Hierbei nimmt die Ladungsdichte am acac-Liganden zu, während sie an den restlichen Liganden und am Platin-Atom verringert wird (siehe Abb. 6). Hier kann ebenfalls von einem LLCT/MLCT-Charakter gesprochen werden. Als wichtigster Übergang stellt sich die Anregung von HOMO nach LUMO+1 mit einem Anteil von 24,03% an der Gesamtwellenfunktion dar.
Abb. 6: Darstellung der Differenzdichten für die relevanten Übergänge im berechneten Absorptionsspektrum. Blaue Flächen entsprechen einer Zunahme der Elektronendichte, rote Flächen stehen für eine Abnahme der Elektronendichte. Links: Anregung von S0 nach S25 (223 nm). Mitte:
Anregung von S0 nach S17 (243 nm). Rechts:Anregung von S0 nach S4 (299 nm).
Im experimentellen Absorptionsspektrum (siehe Abb. 5) liegt der höchste Peak mit einer Intensität von 1 bei einer Wellenlänge von 220 nm. Dieser kann dem höchsten Peak des berechneten Spektrums ohne SOC bei 243 nm zugeordnet werden. Der Peak des berechneten Spektrums bei 223 nm ist in dem experimentellen Spektrum nicht vorhanden. Ebenso kann die Schulter des experimentellen Spektrums bei ca. 245 nm nicht in dem berechneten Spektrum wiedergefunden werden. Auffällig ist, dass in dem experimentellen Spektrum ab einer Wellenlänge von 290 nm viele kleinere Banden auftreten. Die ersten beiden Banden bei 290 nm und 305 nm können vermutlich dem Peak bei 299 nm im berechneten Spektrum zugeordnet werden. Die restlichen drei Peaks bzw. Schultern im Experiment ab etwa 330 nm werden wahrscheinlich durch die Schulter im berechneten Spektrum ohne SOC bei 345 nm beschrieben.
Durch die Einschaltung der Spin-Bahn-Kopplung kommt es zu einer Blauverschiebung des berechneten Spektrums. Der Peak bei 223 nm im berechneten Spektrum ohne SOC taucht im berechneten Spektrum mit SOC nicht auf, da dieses Spektrum erst ab einer Wellenlänge von 220 nm beginnt. Hier muss festgestellt werden, dass die Anzahl der in der SOC-Rechnung verwendeten Wurzeln nicht ausgereicht hat, um den Wellenlängenbereich unter 220 nm wiederzugeben. Die Kurvenverläufe der beiden berechneten Spektren sind sich ansonsten qualitativ sehr ähnlich. Es kommt durch den Einfluss der Spin-Bahn-Kopplung zu einer Verkleinerung der Intensität des dritten Peaks aus der Rechnung ohne SOC, während die Intensität der Schulter aus der Rechnung ohne SOC angehoben wird. Insgesamt wird durch Einbezug der Spin-Bahn- Kopplung in den Rechnungen eine Annäherung an das experimentelle Absorptionsspektrum beobachtet. Dies lässt sich dadurch erklären, dass die Peaks im Spektrum unter anderem durch MLCT-Übergänge unter Beteiligung des Platinatoms verursacht werden.
Es muss weiterhin beachtet werden, dass Umgebungseffekte sich auf den Verlauf der Absorptionsspektren auswirken. Das experimentelle Absorptionsspektrum wurde in
Umgebungseffekte berücksichtigt wurden. Die kleineren Banden im experimentellen Spektrum ab 290 nm, die so in den berechneten Spektren nicht vorhanden sind, sind möglicherweise auf Umgebungseinflüsse zurückzuführen.
5.1.3 Der S1-Zustand
Für den Vergleich der Geometrieparameter werden hier und im Folgenden alle Bindungslängen sowie die Bindungswinkel am Platin und ausgewählte Diederwinkel, die für eine Untersuchung der Planarität der Geometrien bedeutsam sind, zugrunde gelegt (siehe Anhang Tabellen A1 - A3). Als Diederwinkel wurde jeweils der Winkel zwischen dem NHC- und dem DBF-Liganden (C2-N2-C11-C1), der Winkel zwischen dem acac-Liganden und der Pt-NHC-Bindung (C6-O2-Pt-C2), der Winkel zwischen dem acac-Liganden und der Pt-DBF-Bindung (C3-O1-Pt-C1) sowie der Winkel der Liganden zueinander (C2-C1-O1-O2) ausgewählt.
Die optimierte S1-Geometrie unterscheidet sich nur geringfügig von der S0-Geometrie.
Komplex 1 liegt in der S1-Geometrie ebenfalls annähernd planar vor.
Die größte Abweichung zwischen den Bindungslängen beider Geometrien liegt zum einen in der Verlängerung der C11-C1-Bindung am DBF-Liganden und zum anderen in der Verkürzung der Bindung zwischen dem DBF- und dem NHC-Liganden (C11-N2) um jeweils 6 pm. Dieser Unterschied kann durch Betrachtung der Differenzdichten erklärt werden (siehe Abb. 7). Entlang der C11-C1-Bindung kommt es zu einer Abnahme der Elektronendichte und damit zu einer Bindungsverlängerung, wohingegen die Elektronendichte an der C11-N2-Bindung zunimmt und die Bindung dadurch kürzer wird.
Der größte Unterschied bei den Bindungswinkeln am Platin beträgt 1,1°. Bei den Diederwinkeln fällt auf, dass der acac-Ligand um 3,3° aus der restlichen Ligandenebene herausragt (C2-C1-O1-O2).
Der optimierte S1-Zustand von Komplex 1 liegt mit seiner TDDFT-Energie 3,52 eV über dem S0-Minimum.
Der S1-Zustand wird durch einen Übergang von HOMO nach LUMO mit einem Anteil an der Gesamtwellenfunktion von 89,3 % verursacht. Anhand der Differenzdichten (siehe Abb. 7) ist erkennbar, dass zum größten Teil Ladungsdichte auf den NHC- Liganden übertragen wird. Gleichzeitig nimmt am Platin die Ladungsdichte ab. Der S1- Zustand kann als gemischter LLCT/MLCT-Zustand charakterisiert werden.
5.1.4 Die T1-Zustände
Für Komplex 1 konnten drei T1-Geometrien optimiert werden, deren Zustände unterschiedlichen Charakter aufweisen. Sie werden im Folgenden namentlich unterteilt in einen T1a-, T1b-, und T1c-Zustand. Die Geometrieparameter sind im Anhang zu finden (Tabellen A1 – A3).
5.1.4.1 Der T1a-Zustand
Die T1a-Geometrie ist ebenfalls relativ planar, und im Vergleich zu der S0-Geometrie liegen auch hier nur marginale Abweichungen in den Geometrieparametern vor. Es gibt nur eine deutliche Vergrößerung der Bindungslänge bei der C11-C1-Bindung des DBF- Liganden von 8 pm, welche ebenfalls durch eine Abnahme der Elektronendichte an dieser Bindung erklärt werden kann (siehe Abb. 7), während bei den betrachteten Bindungswinkeln die größte Differenz bei der C2-Pt-C1-Bindung mit 0,9° liegt. Die Diederwinkel sind annähernd identisch mit denen aus der S0-Geometrie.
Der optimierte T1a-Zustand liegt mit seiner TDA-Energie 2,74 eV über dem S0- Minimum.
Der T1a-Zustand wird wie der S1-Zustand durch einen HOMO-LUMO-Übergang verursacht, wobei der Anteil an der Gesamtwellenfunktion mit 92,4% hier sogar etwas größer ist. Bei der Betrachtung der Differenzdichten (siehe Abb. 7) wird deutlich, dass Elektronendichte innerhalb des NHC– und DBF-Liganden umverteilt wird, während die Ladungsdichte am Platin abnimmt. Dieser Zustand besitzt deswegen LC/MLCT- Charakter.
Abb. 7: Differenzdichten des S1-Zustandes (links) und des T1a-Zustandes (rechts).
5.1.4.2 Der T1b-Zustand
In der T1b-Geometrie ist die Planarität, die in den vorherigen optimierten Strukturen näherungsweise vorhanden war, aufgehoben. Die Kohlenstoffkette am acac-Liganden ist nach oben hin „abgeknickt“ (siehe Abb. 8). Die Diederwinkel zwischen dem acac- Liganden und der Pt-NHC- bzw. Pt-DBF-Bindung betragen 148,1° bzw. 146°. Bei den Bindungswinkeln liegt im Vergleich zu der T1a-Geometrie die größte Abweichung bei der C2-Pt-C1-Bindung mit einer Differenz von 0,9°. Im Vergleich zur S0-Geometrie beträgt die größte Abweichung 0,8° bei der C1-Pt-O1-Bindung.
Bei den Bindungslängen liegen verglichen mit der S0-Geometrie keine größeren Unterschiede vor. Ein markanter Unterschied zu der T1a-Geometrie tritt wiederum bei der C11-C1-Bindung des DBF-Liganden auf, die in der T1b-Geometrie um 8 pm kürzer ist.
Für den optimierten T1b-Zustand ergibt sich mit TDA eine adiabatische Energiedifferenz von 3,00 eV relativ zum S0-Minimum.
Den größten Anteil am T1b-Zustand besitzt ein HOMO-1-LUMO-Übergang mit einem Anteil an der Gesamtwellenfunktion von 52,2%. Anhand der Differenzdichten (siehe Abb. 8) ist erkennbar, dass beim T1b-Zustand zum größten Teil Ladungsdichte innerhalb des acac-Liganden verschoben wird, während die Elektronendichte am Platin abnimmt.
Der T1b-Zustand besitzt deswegen ILCT/MLCT-Charakter. Dieser größtenteils ligandenzentrierte Zustand ist die Ursache für das „Abknicken“ des acac-Liganden.
Des Weiteren liegen noch Übergänge mit einem geringeren Anteil an der Gesamtwellenfunktion vor. Zum einen tauchen noch ein HOMO-LUMO-Übergang mit einem Anteil von 21,1% sowie ein HOMO-2-LUMO-Übergang mit einem Anteil von 11,2% auf.
5.1.4.3 Der T1c-Zustand
In der T1c-Geometrie liegt der acac-Ligand verdreht zum restlichen Ligandengerüst vor und die annähernd quadratisch-planare Koordination des Platin-Atoms ist aufgehoben (siehe Abb. 8). Die größten Abweichungen im Vergleich zu den anderen optimierten Strukturen finden sich dementsprechend in den Bindungsverhältnissen am Platinatom.
Die Bindungen am Platin sind aufgrund der Verzerrung der Struktur länger. So liegt die größte Abweichung in den Bindungslängen im Vergleich zur T1a-Geometrie bei der Pt-
Unterschied findet sich auch in der Pt-O2-Bindung am acac-Liganden, welche von der T1a- zur T1c-Geometrie um 9 pm größer wird.
Durch die Verdrillung des acac-Liganden wird der C1-Pt-O1-Winkel stark aufgespreizt.
So vergrößert sich dieser Winkel von der T1a- zur T1c-Geometrie um 11,2°. Der acac- Ligand ragt um 63,9° (C2-C1-O1-O2) aus dem restlichen Ligandengerüst heraus.
Aus der Verdrillung resultiert ein Diederwinkel von 174° zwischen dem acac-Liganden und der Pt-NHC-Bindung. Des Weiteren beträgt der Diederwinkel zwischen dem acac- Liganden und der Pt-DBF-Bindung 83,6°.
Der optimierte T1c-Zustand liegt mit seiner TDA-Energie 2,83 eV über dem S0- Minimum. Damit befindet der T1c-Zustand für Komplex 1 energetisch zwischen dem T1b- und dem T1a-Zustand.
Der T1c-Zustand ist durch einen HOMO-LUMO-Übergang mit einem Anteil von 95,2%
an der Gesamtwellenfunktion charakterisiert. Bei Betrachtung der Differenzdichten (siehe Abb. 8) wird deutlich, dass es sich um einen MC-Zustand handelt, bei welchem es zu einer dd*-Anregung am Platinatom kommt. Dieser metallzentrierte Charakter ist die Ursache für die Verzerrung der Bindungsverhältnisse am Platinatom und die damit einhergehende Verdrehung des acac-Liganden aus der restlichen Ligandenebene.
Abb. 8: Frontalansicht der T1b-Geometrie (oben links) mit den Differenzdichten des T1b-Zustandes (oben rechts) und Frontalansicht der T1c-Geometrie (unten links) mit den Differenzdichten des T1c-Zustandes (unten rechts).
5.1.5 DFT/MRCI-Rechnungen
Auf der Grundlage von DFT/MRCI-Rechnungen an den optimierten Geometrien wurde die energetische Lage der Zustände der optimierten Strukturen untersucht. Hierbei wurden die Energien relativ zur S0-Energie auf DFT/MRCI-Niveau an der S0-Geometrie berechnet. Die energetische Lage der Zustände ist in Abb. 9 graphisch dargestellt.
Abb. 9: Graphische Darstellung der energetischen Lage der Zustände der optimierten Geometrien an der jeweiligen optimierten Geometrie relativ zum S0-Zustand am S0-Minimum.
Anhand von Abb. 9 fällt auf, dass der T1a- und T1b-Zustand an der S0-Geometrie verglichen mit ihrer Lage an den anderen Geometrien mit 2,99 eV bzw. 3,09 eV energetisch sehr nah beieinander liegen.
An der S1 und T1a-Geometrie liegen der S1- und der T1b-Zustand mit einem Unterschied von 0,08 eV bzw. 0,05 eV energetisch fast auf dem gleichen Niveau. Mit Ausnahme der S1-Geometrie sind der T1a- und T1b-Zustand an allen Geometrien energetisch unter dem S1-Zustand zu finden. Der T1c-Zustand stellt abgesehen von der T1c-Geometrie an allen anderen Geometrien in dieser Betrachtung den höchsten Zustand dar. An der T1c- Geometrie ergibt sich im Vergleich zu den restlichen Geometrien ein anderes Bild. Der T1c-Zustand wird energetisch abgesenkt und ist, verglichen mit den Zuständen an den restlichen Geometrien, mit 2,71 eV der am tiefsten liegende Triplett-Zustand. Die restlichen Zustände werden an der T1c-Geometrie energetisch stark angehoben. Ist an
0 1 2 3 4 5 6 7
S0 S1 T1a T1b T1c
optimierte Geometrien
Energetische Lage der Zustände im DFT/MRCI für Komplex 1
S0 S1 T1a T1b T1c
betrachteten Triplett-Zustand noch sehr groß, so verringert sich dieser Abstand an der T1c-Geometrie bedeutend. Er beträgt an dieser Geometrie nur noch 0,46 eV.
5.1.6 Emissionsspektren
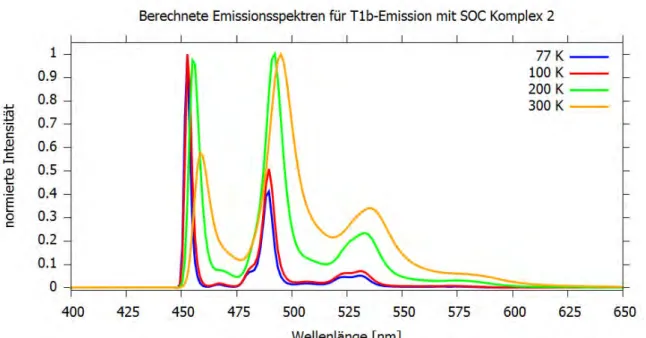
Für den nach TDA energetisch am niedrigsten liegenden Triplett-Zustand, den T1a- Zustand, wurden temperaturabhängige Franck-Condon-Emissionsspektren berechnet.
Hierbei wurde eine adiabatische Energiedifferenz nach DFT/MRCI zwischen dem S0- Zustand an der S0-Geometrie und dem T1a-Zustand an der T1a-Geometrie von 22322 cm-1 zugrunde gelegt. Aus den SOCI (engl.: Spin-Orbit Configuration Interaction)- Rechnungen von Frau Prof. Marian ergibt sich eine adiabatische Energiedifferenz von 23138 cm-1 zwischen dem S0-Zustand an der S0-Geometrie und dem T1a-Zustand an der T1a-Geometrie.
Die experimentellen Emissionsspektren wurden bei einer Komplex-Konzentration von 2 Massen-% in einem PMMA-Film bei Umgebungstemperaturen von 5 K, 77 K, 100 K, 200 K und 300 K aufgenommen.[5] Für die Berechnung der Emissionsspektren wurden Temperaturen von 77 K, 100 K, 200 K und 300 K gewählt. Es hat sich herausgestellt, dass bei einer Temperatur von 200 K experimentelles und berechnetes Emissionsspektrum am besten übereinstimmen. In Abb. 10 sind die berechneten Spektren mit und ohne Einfluss der Spin-Bahn-Kopplung bei 200 K sowie das experimentelle Spektrum bei 200 K dargestellt. Die Spektren sind auf eine Intensität von 1 normiert. Der Wellenlängenbereich entspricht dem des experimentellen Spektrums.
Abb. 10: Berechnetes Emissionsspektrum (rot), berechnetes Emissionsspektrum mit Spin-Bahn- Kopplung (violett) und das experimentelle Emissionsspektrum (blau) bei einer Temperatur von 200 K.
In den berechneten Spektren liegen drei Peaks vor (siehe Abb. 10). Der intensivste Peak beim berechneten Spektrum ohne SOC, der auf eine Intensität von 1 normiert ist, liegt bei einer Wellenlänge von 473 nm. Der zweite Peak mit einer relativen Intensität von 64% ist bei einer Wellenlänge von 513 nm zu finden. Der dritte und kleinste Peak bei einer Wellenlänge von 553 nm besitzt eine relative Intensität von 9%. Die Berechnungen zeigen, dass eine Schwingung bei 1656 cm-1 existiert, die auf dem DBF- und NHC-Liganden lokalisiert ist und das größte Displacement aufweist. Anhand des berechneten Spektrums bei 77 K, welches im Wellenzahlenbereich geplottet und bei dem der erste und intensivste Peak auf den Ursprung verschoben wurde (siehe Anhang Abbildung B21), kann nachvollzogen werden, dass der energetische Abstand zwischen den drei intensivsten Banden, die den Banden des berechneten Spektrums bei 200 K entsprechen, bei ungefähr 1660 cm-1 liegt, was für den Charakter einer Schwingungsprogression spricht. Dieser Wert gehört vermutlich zu der Schwingung von 1656 cm-1. Zu beachten ist, dass beim Spektrum bei 77 K im Vergleich zum Spektrum bei 200 K die letzte Bande sich in zwei Banden aufspaltet und bei der Ermittlung des energetischen Abstands die letzte der beiden Banden ausgewählt wurde.
Im experimentellen Emissionsspektrum sind zwei Peaks sowie eine Schulter zu erkennen. Die erste Bande mit der höchsten Intensität bei 463 nm ist mit der ersten Bande aus dem berechneten Spektrum vergleichbar. Die zweite Bande bei 497 nm mit einer relativen Intensität von 50% entspricht der zweiten Bande aus der Rechnung,
wobei die Intensität der zweiten berechneten Bande um 14 % größer ist. Die Schulter des experimentellen Spektrums bei 531 nm mit einer Intensität von 14% relativ zur größten Bande kann der dritten Bande des berechneten Spektrums zugeordnet werden, wobei die relative Intensität der Schulter etwas höher liegt.
Die Energiedifferenz im experimentellen Spektrum zwischen erstem und zweitem Peak und zweitem und drittem Peak beträgt 1478 cm-1 und 1288 cm-1. Qualitativ betrachtet sehen sich die Verläufe des berechneten und des experimentellen Spektrums sehr ähnlich. Das berechnete Spektrum ohne SOC ist im Vergleich zum experimentellen Spektrum rotverschoben.
Durch den Einfluss der Spin-Bahn-Kopplung ergibt sich eine um 816 cm-1 größere adiabatische Energiedifferenz. Dies führt zu einer Blauverschiebung des berechneten Spektrums. Der Abstand der Peaks zwischen berechnetem Spektrum und experimentellem Spektrum verringert sich. Ähnlich wie beim Absorptionsspektrum kann unter Berücksichtigung der Spin-Bahn-Kopplung eine Annäherung an das experimentelle Emissionsspektrum beobachtet werden.
Für den T1b-Zustand wurde kein Emissionsspektrum berechnet, da dessen Geometrie mit einem nach oben „abgeknickten“ acac-Liganden stark von der annähernd planaren Geometrie des S0-Zustandes abweicht. In diesem Fall würde die harmonische Näherung, die der Berechnung des Franck-Condon-Emissionsspektrums zugrunde liegt, versagen und man würde ein stark verbreitertes Spektrum erhalten.
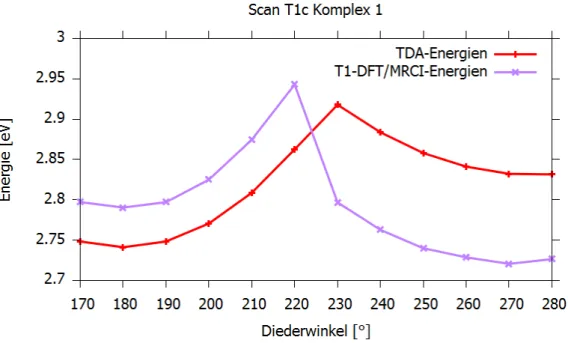
5.1.7 Scan von der T1a- zur T1c-Geometrie
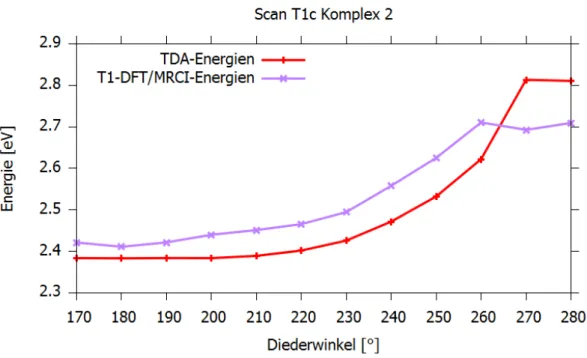
Zur Ermittlung der Energiebarriere zwischen dem nach TDA energetisch am niedrigsten liegenden Triplett-Zustand (T1a) und dem T1c-Zustand, der einen MC-Zustand darstellt, wurde ausgehend von der T1a-Geometrie ein Scan entlang des Diederwinkels C3-O1-Pt- C1 durchgeführt. Mit einem Ausgangspunkt von 170° wurde dieser Winkel in 10°- Schritten bis zu einem Endpunkt von 280°, der in etwa dem Diederwinkel der T1c- Geometrie entspricht, vergrößert. An jedem Schritt wurde mit TDA in den ersten Triplett-Zustand angeregt und optimiert, wobei der Diederwinkel eingefroren wurde.
Anschließend wurden an jeder optimierten Struktur DFT/MRCI-Rechnungen mit 10 Wurzeln für die Triplett-Zustände durchgeführt. In Abb. 11 sind die T1-DFT/MRCI- und TDA-Energien der Geometrien gegen den Diederwinkel C3-O1-Pt-C1 aufgetragen.
Die T1-DFT/MRCI-Energien sind relativ zur Energie des S0-Zustandes am S0-Minimum