Totalsynthese von Belizentrin Methylester
199
0
0
Volltext
(2)(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)(11)
(12)
(13)
(14)(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
(42)
(43)
(44)
(45)
(46)
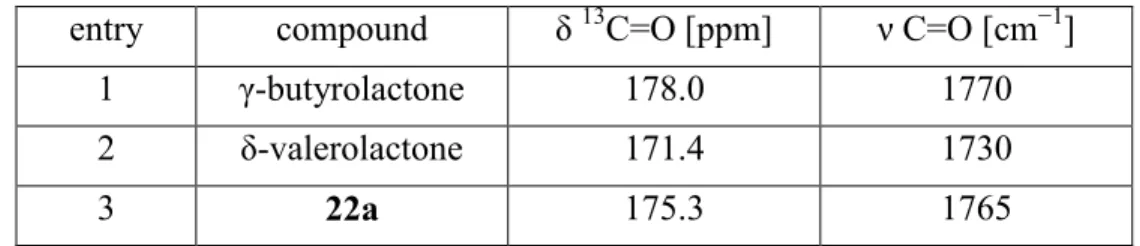
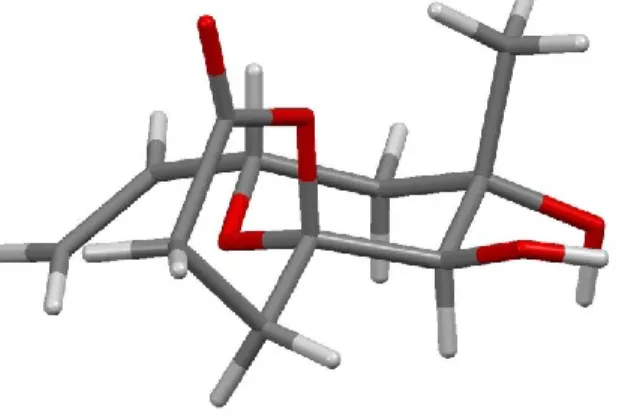
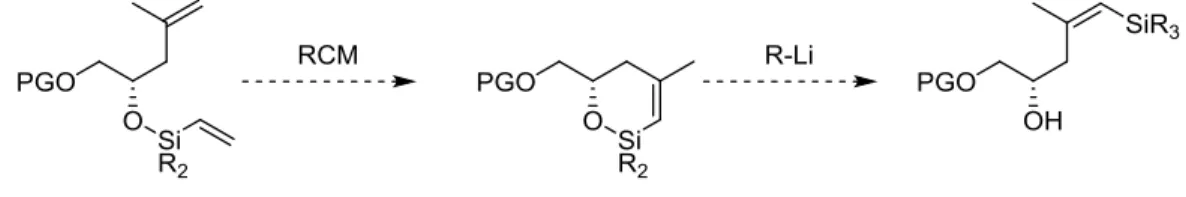
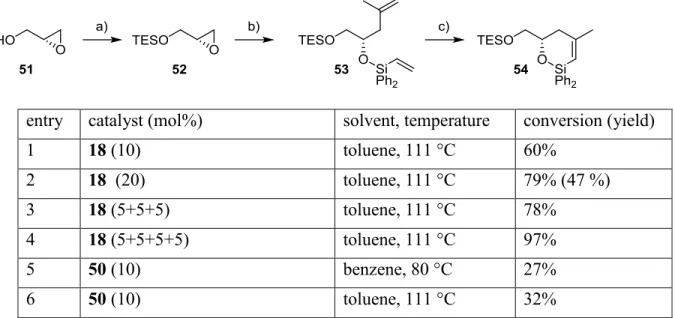
Abbildung
+7


ÄHNLICHE DOKUMENTE
The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure.. To the resulting mixture, tert-butylalcohol,potassiumderivative (14.04 mmol)
The combined organic layers were washed with distilled water, dried over Na 2 SO 4 and the solvent evaporated under reduced
The combined organic layers were washed with water (20 mL) and saturated aqueous sodium chloride solution (20 mL), the washed solution was dried over sodium sulfate and the
The layers were separated and the aqueous layer was extracted with ethyl acetate (3×30 mL), the combined organic layers were washed with saturated aqueous
combined organic layers were washed with aqueous HCl (1 M, 5 mL) and brine (5 mL). The organic solution was dried over Na 2 SO 4 and the solvent was evaporated under reduced..
The aqueous layer was extracted with diethyl ether (2x100 mL), the combined organic extracts were dried over sodium sulfate and all volatiles were removed under reduced pressure.
The organic phase was separated, washed with a saturated aqueous NH 4 Cl solution (10 mL), dried over an- hydrous MgSO 4 and concentrated under reduced pressure.. The residue
The organic layer was washed with saturated sodium chloride solution (100 mL), dried (MgSO 4 ) and concentrated under reduced pressure to give 12 as colourless oil...
