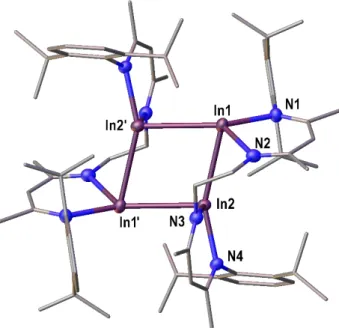
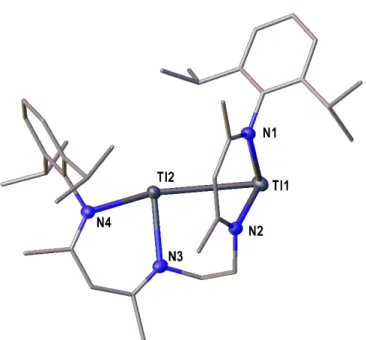
Polynuclear Group 13 Compounds: Synthesis, Characterization, and Reactivity
255
0
0
Volltext
(2)(3)
(4)(5)(6)
(7)(8)
(9)(10)
(11)(12)
(13)(14)(15)
(16)
(17)
(23)
(29)
(32)
(33)
(34)
(35)
(36)
(37)
(38)(39)
(40)(41)
(42)
Abbildung
+7
ÄHNLICHE DOKUMENTE
Although several syntheses of borazine derivatives using borazine as starting material are reported, bor- azine is not a suitable starting material for further syntheses due to
There are four inequivalent pro- tons and six inequivalent carbon atoms (some of them broadened) of the η 6 -bound cymene ligand giving res- onances in the 1 H resp. 13 C NMR
Institut f¨ur Anorganische Chemie, Julius-Maximilians-Universit¨at W¨urzburg, Am Hubland, 97074 W¨urzburg, Germany. Reprint requests
The aforementioned spectro- scopic data as well as the required twofold excess of BeCl 2 already indicate that [Pd(PCy 3 ) 2 ] does not form a Lewis base adduct with BeCl 2 in
A detailed analysis of the J 1 coupling taking into account the molecular structures of the three available heptanuclear com- plexes [Mn III 6 M III ] 3 + + + (M = Cr, Fe, Co)
As the chosen coupling scheme did not provide a reasonable reproduction of the experimental data, our next approach was to take into account a coupling of Mn III ions belonging
The Kamlet-Taft solvatochromic comparison method was used to separate and quantify these effects: An increase in the percentage of organic co- solvent in the medium enhances
This thesis focuses on the development of molecular mechanical (MM) methods and force fields to model hyper-valent molecules, transition metal complexes and ultimately, the study
