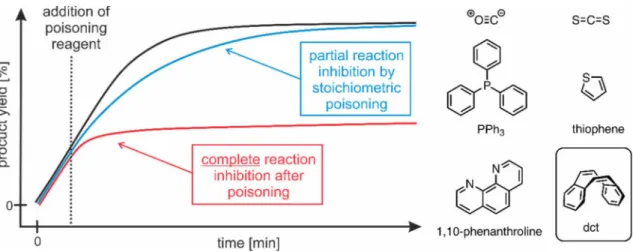
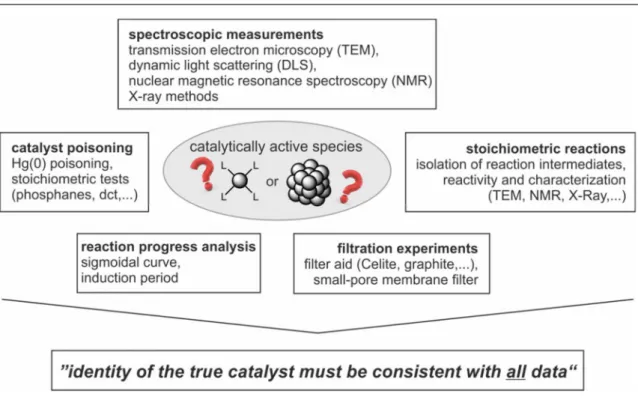
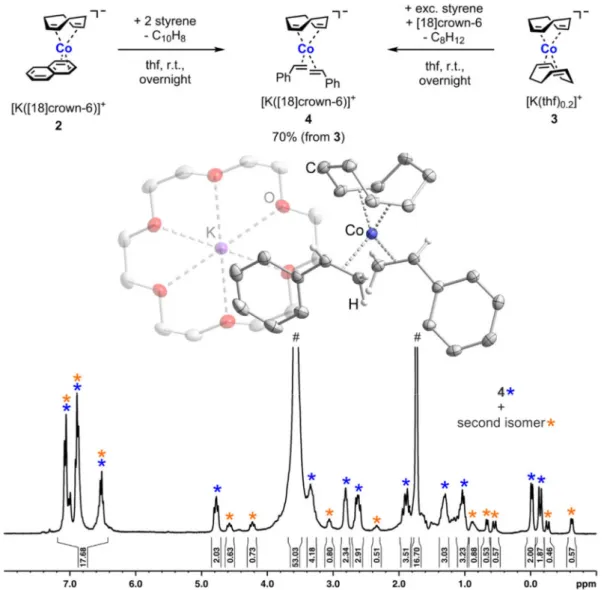
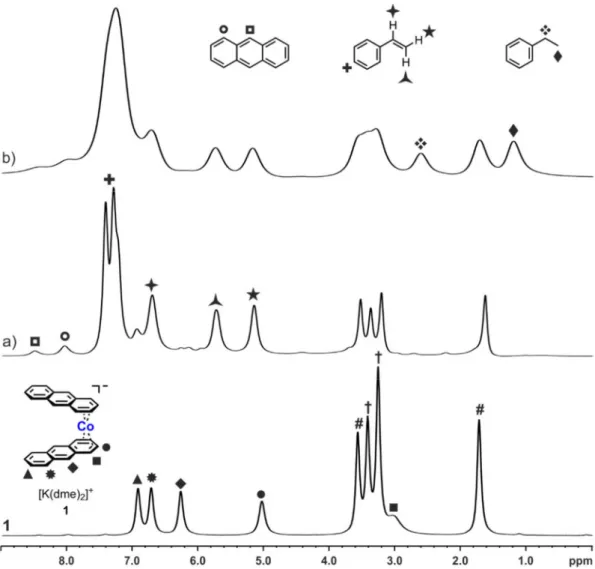
ANOPARTICLES AS C ATALYSTS N E VALUATION OF C OBALTATES AND C OBALT N OBALT -C ATALYZED H YDROGENATIONS : A H OMOGENEOUS VS . H ETEROGENEOUS C
179
0
0
Volltext
(2)(3)
(4)(5)
(6)(7)
(8)
(9)
(10)
(11)
(12)(13)
(14)(15)
(16)
(17)
(18)
(19)
(20)
(21)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
Abbildung

+7

ÄHNLICHE DOKUMENTE
Sondern heut ist der Kamps in j^edes einzelnen Menschen B ru st verlegt : D a ist heut keiner mehr, der nicht christliche Gedanken in sich trüge, auch wenn
Darin hatte er unter anderem Homosexualität als eine „Degenerationsform der Gesellschaft“ bezeichnet und gesagt: „Diese Homolobby, dieses Teuflische kommt immer stärker,
Duck, chicken and beef with Shanghai Paksoi, Bell pepper, broccoli and sweet peas fried in a
Wir haben ja schliesslich im Gegensatz zur Zeit der Spanischen Grippe, unsere hochtechnisierte „Medizin“ und auch ist die Sauberkeit und Hygiene mit der von Anfang des
2 Wenn die Reaktion zu exotherm wird (starkes Sieden des Ethers) muss mit einem Eisbad gekühlt und die Zutropfgeschwindigkeit reduziert werden.. Nach Zugabe des Alde- hyds wird
The most common way to achieve site-selectivity in direct C – H bond activation on arenes is the use of a directing group, which is usually placed in the ortho-position to the C –
che zu bezweifeln hat: so beweiset es, in welcher guten Meinung Pattkul seines Wandels wegen bey andern gestanden haben müsse. Des Churfürsten Johann Georg des
In ein e m zweiten Schritt werden sozio-politische und ökonomische Bedingungen der Entwicklung, Anwendung und Folgebearbeitung unterschiedlicher Nutzungsformen der
