Crosstalk between membrane trafficking and cell adhesion: The role of the SNARE protein TI-VAMP in neuronal morphogenesis
131
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
Abbildung

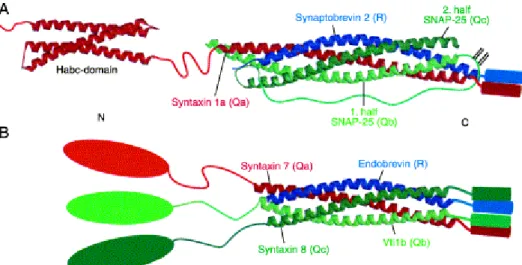


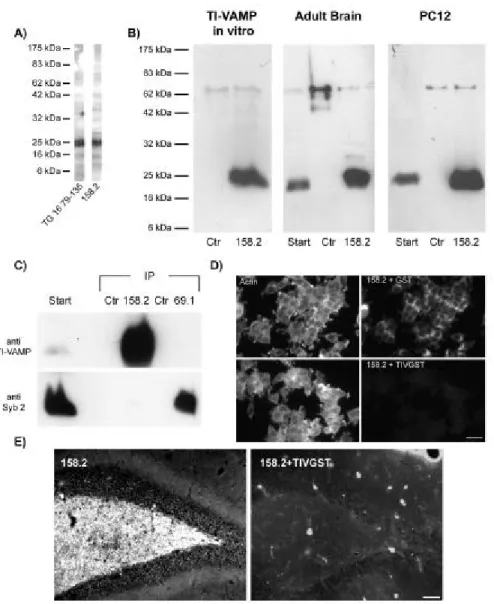
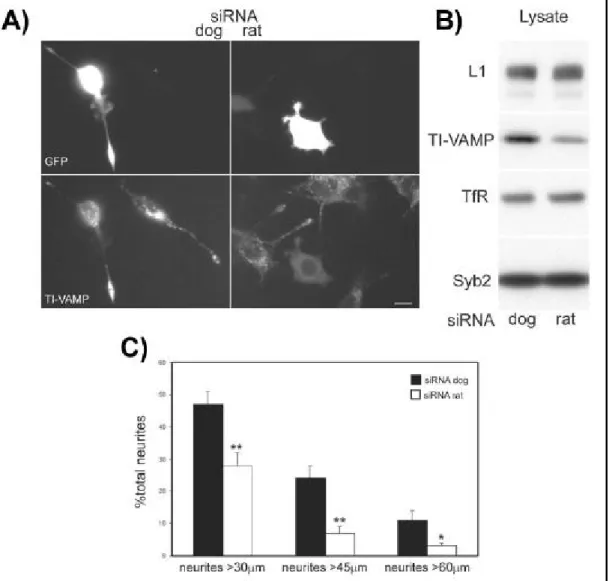
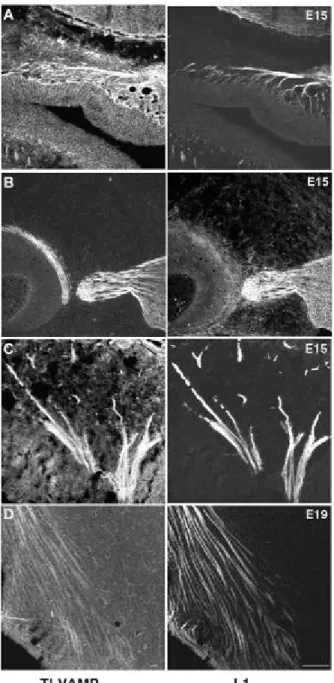
+7



ÄHNLICHE DOKUMENTE
A recent study using electron paramagnetic resonance has reported that the association of Munc18-1 with the syntaxin1a/SNAP25a complex can result in the formation of a complex
Comment on: Folkecology, Cultural Epidemiology, and the Spirit of the Commons, by Scott Atran et al.
Atran and associates conclude that Q’eqchi’ who moved into central Pete´n around the 1970s seem unable to adapt to the lowland environment or learn from their neighbors because of
: Graph illustrating the Dendritic Atrophy phenotype in the mice deficient in Cap23 and comparison with wild type mice Wild type mice (dark line), Cap23 heterozygotes (pink line)
The Skp1-Cullin1-F-box protein (SCF) E3 ubiquitin ligase and in particular the substrate-recruiting adaptor subunit F-box proteins have emerged as essential mod- ulators of
TABLE 1 Average and maximum C stocks in living and dead volumes for forest registered as managed and unmanaged in Germany, based on plot data from the national forest
The left example tests how well different peak detection methods can identify peaks in synthetically generated data.. The right example is an experimental benchmark data set of
The left example tests how well different peak detection methods can identify peaks in synthetically generated data.. The right example is an experimental benchmark data set of
The Poisson distribution expresses the probability of a given number of events occurring in a fixed in- terval of time or space if these events occur with a known constant mean rate
