Multivalently binding antagonists for the neurokinin-1 receptor
DISSERTATION ZUR ERLANGUNG DES DOKTORGRADES DER NATURWISSENSCHAFTEN (DR. RER. NAT.) DER FAKULTÄT
CHEMIE UND PHARMAZIE DER UNIVERSITÄT REGENSBURG
vorgelegt von Anika Veser aus Ravensburg
im Jahr 2018
This work was carried out from November 2012 until November 2018 at the Department of Pharmaceutical Technology of the University of Regensburg.
The thesis was prepared under supervision of Prof. Dr. Achim Göpferich.
Submission of the PhD application: 26.11.2018
Date of examination 01.02.2019
Examination board: Chairman: Prof. Dr. Jens Schlossmann
1. Expert: Prof. Dr. Achim Göpferich
2. Expert: Prof. Dr. Joachim Wegener
3. Expert: Prof. Dr. Rainer Müller
To my family
der Funken will.
Johann Gottfried von Herder
Table of Contents
Chapter 1 Introduction ... Page 1
Chapter 2 Goals of the thesis ... Page 19
Chapter 3 Identification of target cell candidates with
neurokinin-1 receptor expression ... Page 23 Chapter 4 Multivalently binding quantum dots for
neurokinin-1 receptor targeting ... Page 49 Chapter 5 The challenge of site-specific ligand PEGylation in the
synthesis of multivalently binding drugs ... Page 73 Chapter 6 Aprepitant, a small molecular weight antagonist for the
neurokinin-1 receptor and its functionalization for
nanoparticle coupling ... Page 107 Chapter 7 Enzymatic ligand activation on the surface of nanoparticles ... Page 125 Chapter 8 Summary and Conclusions ... Page 155
Appendix ... Page 161
Abbreviations ... Page 162
Curriculum Vitae ... Page 166
Acknowledgements ... Page 167
Chapter 1
Introduction
The interrelation of inflammation and pain
The body´s homeostasis is regulated by three well-connected systems: the nervous system, the endocrine system, and the immune system. Direct cell-to-cell contact, soluble signaling molecules like neuropeptides, hormones, and cytokines together provide effective and rapid channels for mutual communication [1]. For this reason, inflammation and pain are closely related and the severity of both correlate to each other (Figure 1).
Figure 1: Relationship between innervation, inflammation, vascularization, damage, and pain.
This neuroimmune interplay is bidirectional.
Based on this relationship, if inflammation regresses, pain symptoms should also be relieved.
In contrast, in autoimmune diseases, the state of inflammation and pain is ongoing. This phenomenon which is attributed to neuroinflammation, also called neurogenic inflammation.
This kind of inflammation is characterized by the release of inflammatory substances from peripheral endings of primary afferent nerve fibers, which seem to be responsible for preserving the status of inflammation and pain in an unending cycle. These inflammatory substances are categorized as neuropeptides [2]. These peptides are able to interfere between the central nervous system (CNS) and the periphery because they are released from nerves in the CNS and from peripheral nerves, endothelial tissues, and various immune cells, including T and B lymphocytes, monocytes/macrophages, neutrophils, and mast cells [3, 4].
The combination of up-regulation of expression of these substances and their receptors in
the pathophysiological state of an autoimmune disease, together with further factors like the
release of cytokines, appears to cause an inflammatory state. Inflammation may include
clinical symptoms such as heat, redness, swelling, pain, and decreased function, symptoms that significantly reduce the quality of life of affected patients [5]. Neuropeptides, like substance P, bradykinin, or calcitonin gene-related peptide (CGRP) induce vasodilatation and plasma extravasation of proteins to the site of injury, which causes swelling. Substance P is a sensory neuropeptide. Ulf von Euler and John Gaddum identified substance P in 1931;
they concentrated it in a powdered form and therefore named it substance P [6].
In the case of rheumatoid arthritis (RA), the volume of synovial fluid in the joint cavity is increased compared to the unaffected joint, due to the presence of neuropeptides.
Osteoarthritis (OA) may develop in later stages, which is characterized by cartilage breakdown, the outgrowth of bone, bone sclerosis, and synovitis [5]. In contrast to RA, OA is not defined as an inflammatory disorder because the leukocyte number in the synovial fluid is below the typical threshold of 2000 cells/mm
3[7]. However, in both disease states, patients feel pain because of the activation of primary afferent nociceptors, which innervate the joint´s synovial membrane and are connected to the spinal cord for transmitting the received stimuli to higher brain centers where the perception of pain occurs. In contrast to the synovium, healthy cartilage does not contain blood vessels and is therefore a hostile area for a deep innervation by nerve fibers. The perivascular localization of nerves implies that vascularization is the driver behind innervation [8]. With increasing cartilage destruction, vascularization and innervation increase and is therefore an indicator for severity and may be a potential source of pain, particularly in patients with OA.
Arthritic pain affects millions of people worldwide, but understanding of the origin of the painful sensation is still limited. Current understanding is that during inflammation, the joint nerves become sensitized to mechanical stimuli through the actions of neuropeptides, eicosanoids, activated receptors, and ion channel ligands [9].
That neuropeptide antagonists have anti-inflammatory characteristics is well proven, but
whether they can act as analgesics is controversial and has only be confirmed in experiments
with animals, not with humans. It is suggested that in particular pain states, antagonists
against the neurokinin-1 receptor (NK
1R) may be effective analgesics or at least may be used
as adjuvants to existing analgesics [10]. Many NK
1R antagonists have failed trials of a
variety of clinical pain states, although they have shown in preclinical studies that they can
attenuate nociceptive responses. However, they have not reached the efficacy of typical nonsteroidal anti-inflammatory drugs, or NSAIDs, which are directed against the biosynthesis of prostaglandins by cyclooxygenase, or COX inhibition [10, 11]. In rats, it has been shown that the presence of NK
1R in the knee joint cavity contributes to the induction of pain response, but seems not to be involved in the maintenance of arthritic pain [12]. It is also not clear which neuropeptides are involved in immune response and pain enhancement.
Some believe that it is not substance P that increases inflammation and hyperalgesia, but hemokinin-1, which is also able to bind to several neurokinin receptors with very high affinity to NK
1Rs [13, 14]. Because of its low receptor selectivity compared to substance P, this aspect could be a further reason that selective NK
1R blockers do not reach the desired anti-inflammatory and analgesic effects. In contrast, substance P and glutamate together have been found to contribute to arthritic pain and joint swelling. This concept was proven in an arthritic rat model by co-administration of respective substance P and glutamate antagonists to common dexamethasone therapy [15]. It seems that NK
1R is one component, but not the only key component at the interface of immune and pain responses.
Peripheral targeting of substance p/ NK
1R interactions
The therapeutic potential of targeting substance P and NK
1R interactions in peripheral
inflammatory and CNS-associated disorders and their impact on the pharmacology of pain
are a focus of research [16, 17]. One cornerstone is the observation that NK
1Rs are expressed
on common immune cells and cells of the brain with immune cell-like properties, known as
glia cells and astrocytes. The major ligand of NK
1R, substance P, is mainly stored together
with CGRP and other transmitters in dorsal root ganglion cells of the spinal cord. Upon
depolarization, sensory nerve fiber terminals can quickly and ubiquitously release both
neuropeptides throughout the body´s peripheral tissues. This release is triggered by various
proinflammatory factors [18]. Substance P and CGRP are responsible for either peripheral
neurogenic inflammatory responses or equivalent responses in the CNS by cell activation
via their respective surface receptors. The NK
1R target cells can be either vascular or non-
vascular endothelial cells, various immune cells, or other cell types. Together with cytokines,
overexcited substance P/NK
1R interactions can initiate and promote some autoimmune
diseases, e.g., RA and inflammatory bowel disease, as well as pathogen derived or sterile
neuroinflammatory disorders, e.g., meningitis or multiple sclerosis [17]. The impact of
NK
1R activation is also discussed in studies for a variety of psychiatric disorders, including anxiety and depression [19]. Since substance P crosses the blood-brain barrier (BBB) and is responsible for BBB permeabilization, it seems to be a key factor in the transmission of peripheral inflammatory response to the CNS and therefore a main trigger of autoimmune diseases by activating monocyte-like parenchymal microglia [20]. Like systemic macrophages, microglia cells also express innate immune receptors and have the ability for phagocytosis and therefore represent the brain´s own immune system. However, not only substance P but also cytokines, like interleukins and growth factors, modulate the actions in both defense systems, because they are also produced by microglia [21]. Besides increased cytokine concentrations in the blood and liquor, which is evident during inflammation, it could be found that sensory nerves also have higher neuropeptide content [22]. In parallel, the presence of cytokines regulates NK
1Rs. Figure 2 shows the cross-talk between the CNS and peripheral systems by blood-derived immune cells and peripheral nerve fibers.
Figure 2: Cross-talk between the CNS and various peripheral systems in which the NK1R is upregulated under pathological conditions. Substance P binds to the NK1R by autocrine, paracrine, and/or endocrine mechanisms, and is released from nerve terminals. The neuropeptide reaches the whole body through the bloodstream. This figure is adapted from [23].
Implication of NK
1R expression in cancer and rheumatoid arthritis
The recruitment of immune cells into the joint space is one similarity of RA to the pathology of cancer caused by increased angiogenesis and higher permeability of blood vessels.
Therefore, common strategies for RA treatment exactly pick up regulatory mechanisms in the context of angiogenesis or inflammation for selective interruption. For cancer, some well-known examples are the antibody treatments with bevacizumab or trastuzumab, which both have antiangiogenic, anti-inflammatory, and growth inhibitory effects. In RA, the treatment of choice is also anti-inflammatory, e.g., with etanercept or infliximab to reduce the action of cytokines like TNF-α [24]. Its neutralization leads to the subsequent inhibition of other proinflammatory cytokines. Besides increased cytokine levels, a second similarity between cancer and RA is increased levels of substance P in the blood and in the CNS, and for RA in particular, in the synovial fluid within the joint space. In cancer, substance P is considered a mitogen because it has been associated with tumor growth and metastasis [25].
High cytokine and substance P concentrations are synergistically linked to each other since
the release of substance P from local peripheral nerve fiber terminals is a result of cytokine
stimulation. Substance P then significantly contributes on the one hand to the production of
further cytokines, such as IL-1β, TNF-α and the neutrophil chemoattractant IL-6 by direct
NK
1R interaction on present immune cells. On the other hand, NK
1R interaction on vascular
endothelial cells involves substance P in the recruitment of further immune cells, which
causes higher permeability and vasodilatation [26]. Other proinflammatory effects are the
release of lysosomal enzymes, nitric oxide, and prostaglandin E
2[5, 12]. This increasing
inflammation leads in later stages to tissue destruction and hyperalgesia. There is also strong
evidence that angiogenesis and inflammation are two major driving forces for the progress
of early and later OA stages. Both are closely integrated perpetuating processes, such that
inflammation can trigger angiogenesis, and angiogenesis can speed up inflammation, rather
than cause it, by facilitating the immigration of more inflammatory cells that generate
angiogenic factors [27, 28]. In arthritis, vascular proliferation contributes to the pathogenesis
of synovitis, pannus growth, bone and cartilage destruction, and the formation of
osteophytes [29]. Beside substance P, many more substances are localized or released within
osteoarthritic human synovium, synovial fluid, and articular chondrocytes [30]. One of those
is angiotensin II, which is released from the synovium into the synovial fluid and seems to join with substance P. Several other factors, like various cytokines such as IL-1, IL-4, IL-8, IL-18, and TNF-α are responsible for angiogenesis and may exacerbate inflammation.
However, IL-10 and TGF-β, as well as cytokine inhibitors such as IL-1ra and soluble TNF-R, are also produced as anti-inflammatory counterparts [26]. All these factors were found in increased levels in the synovial fluid from patients with arthritis [29].
Function of different NK
1R isoforms
Accumulating evidence has suggested that NK
1R is closely related to immune responses and may be recognized as a key factor in neuroinflammation. Therefore, NK
1R may represent a potential therapeutic target in the neuro-immune axis. For a better understanding of NK
1R- ligand interactions it is important to know that other neurokinin receptor types exist (NK2R and NK3R) [31], two different isoforms with different intracellular pathways. The broad spectrum of different receptors and their distinct selectivity for certain ligands makes the topic of specific receptor ligand interaction even more complex. The exact functional pathways and the reason for different neurokinin receptor types, different expression profiles, and different ligand affinities, in addition to different NK
1R isoforms with distinct molecular masses is still not clarified. Differences in lengths arise from a lack of 96 amino acids at the intracellular C-terminal end of the receptor, which vary from 407 amino acids for the full-length receptor isoform to 311 amino acids for the truncated receptor isoform [3].
These isoforms originate from two differently spliced mRNAs. It is suggested that the
different receptor versions may have a regulatory role in receptor desensitization, since the
short form has a 10-fold lowered affinity for substance P and is not able to elicit intracellular
G-protein binding, which induces the activation of NFκB via kinase-cascade activation and
eventually results in the production of proinflammatory cytokines [32, 33]. After repeated
induction of these intracellular mechanisms by binding agonists, the full-length receptor
becomes internalized by endosomes. This is not the case for the truncated receptor version
because of the loss of C-terminal Ser/Thr residues, which are essential for receptor
internalization upon G-protein-coupled receptor kinase interaction and β-arrestin
recruitment [34]. The prevalence of different splice variants seems to be distributed into
brain and peripheral tissues, where the full-length form is mostly found in the brain and the
in cancer, the short receptor form is over-expressed and offers a potential target site for anti-substance P and NK
1R treatment [36]. It has been shown recently that the truncated NK
1R also has important functional roles concerning the modulation of immune responses by cytokines and the chemotaxis of macrophages [37].
Multivalent antagonistic NK
1R targeting
Antagonistic treatment of neuropeptide receptors, especially NK
1R, could be a promising
approach to anti-inflammatory pharmacotherapy [1]. The main benefit arises from using a
multivalent technique such as coupling antagonists on the surface of nanoparticles where an
increased effectiveness of treatment by the blockade of multiple receptors at the same time
is expected. This aspect depends on high receptor densities on cell surfaces, which is the
case in neurogenic inflammatory states, where NK
1Rs are over-expressed. The generation
of a spatial proximity of multiple ligands on the surface of the nanoparticle to a highly
receptor crowded cell surface determines whether a receptor blockade is sustained. Keeping
several ligands close to receptors after the initial binding of the first ligand preserves this
proximity. The likelihood for a second ligand binding is relatively high. This state, when
several ligands bind several receptors at the same time, is called multivalency, and the
effect—that an initial receptor binding promotes the binding of a second one—is called
cooperativity. This potent docking principle to cells´ surfaces was found for various
viruses [38]. Other approaches focus on hetero-multivalent binding drugs since single drug
treatments often suffer from limited efficacy and/or high toxicity. Depending on the
nanoparticles´ core material for the design of multivalently acting drugs, cell and organ
toxicity are an issue. For experimental studies, the visibility of particles, homogeneity in
size, and charge are major demands. Quantum dots offer all these benefits and are therefore
a perfect tool to investigate multivalent interactions. Although quantum dots perfectly meet
the specifications for experimental studies, strong evidence suggests cell toxicity in higher
concentrations because of their bright fluorescent cadmium core, and little is known about
QD metabolism [39]. In addition, dense surface PEGylation might increase blood half-life,
and precludes elimination from the body. This results in a large area under the exposure-
time curve and increases the likelihood of toxicity [40]. However, if the concept of
multivalent receptor targeting is proven for a target receptor, there are several possibilities
for exchanging the nanoparticle species.
Peptide and non-peptide NK
1R antagonists
There are two fundamental parameters that determine the binding quality of a drug to its receptor binding site: the affinity and the efficacy. The affinity specifies the propensity of a ligand to form a reversible complex with its receptor. The efficacy describes the intrinsic activity of the ligand, which is the ability to produce a functional response in the form of a signaling cascade in cells and tissues [41, 42]. A full agonist can generate a maximal response and tends to stabilize the active state of the G-protein-coupled receptors, whereas full antagonists have no functional effect. The typical stabilized receptor conformation plays an important role for the resulting intracellular signaling or determining if the receptor is simply blocked [43]. For example, substance P binds to the extracellular loops of the NK
1R, whereas small hydrophobic molecular antagonists like aprepitant bind more deeply between the transmembrane segments III–VII [44] (see Figure 3). Partial agonists induce only a submaximal effect and seem to reduce the rate of G-protein turnover, relative to a full agonist, by strongly stabilizing the ternary complex [45]. In general, partial and full agonists share a certain binding topography, but stabilize distinct receptor conformations [46].
Figure 3: Pattern of 7-transmembrane segments of NK1R. The arrows indicate different binding sites for small molecular lipophilic antagonists and more hydrophilic peptide agonists.
In the case of NK
1R downstream signaling, a full and potent agonist, such as substance P,
would result in intrinsic activity in the form of a great calcium influx into the cytosol even
in picomolar concentrations. Aprepitant is known to act as a full antagonist, which does not
trigger intrinsic activity and can be seen as a receptor blocker. Most full antagonists are
synthetic compounds and therefore do not appear in nature, unlike full agonists like substance P. However, the assumption that synthetic compounds produce no functional response is false. On the contrary, most known synthetic drugs exhibit a range of responses between full agonism and full antagonism and are termed partial agonists because their intrinsic response is reduced compared to their full agonistic analogs. Partial agonists can elicit different receptor responses depending on their environment: In the presence of agonists with higher intrinsic activity, partial agonists can demonstrate antagonist properties by blocking access to the receptor, but on their own they will act as agonists [41]. The design of peptide NK
1R antagonists has focused on the introduction of D-amino acids [47].
However, there are several drawbacks; for example, binding affinities are several orders of magnitude lower than their natural agonists and therefore exhibit poor potency. In addition, partial residual agonist activity and mast cell degranulation activity and neurotoxicity have been observed. Another disadvantage is the inability to discriminate between different tachykinin receptor types [48, 49]. In addition, these peptides are not able to gain access to the CNS through the BBB [50]. Depending on the desired application, though, this aspect may not be a limiting factor except for drugs that explicitly address the brain, such as neurotherapeutics [51]. It can be concluded that peptide as well as non-peptide antagonists are useful for peripheral NK
1R targeting. In the context of this work, both species were selected for in vitro multivalent binding experiments. Spantide I was chosen as the peptide antagonist, and the influence of PEGylation on binding affinity was checked prior to coupling to PEG-coated quantum dots. As the non-peptide antagonist, aprepitant was modified by a short alkyl linker to make it accessible for polyethylene glycol (PEG) coupling.
Cost-benefit balancing of drug PEGylation
Up-to-date PEGylation is one post-synthetic strategy of choice with the aim of increasing
the solubility and circulatory half-lives of sparely soluble and/or enzymatically unstable
drugs. The PEG coatings on the surface of nanoparticles have an additional shielding effect,
which prevents them from aggregation, opsonization, and phagocytosis [52]. The reasons
for the popularity of PEG are simple. Polyethylene glycol is a water-soluble, non-toxic, non-
immunogenic, and commercially available polymer that is approved by the United States
Food and Drug Administration and sold with various functional groups at the polymer chain
endings for individual arbitrary chemical modification by customers [53, 54]. But the challenges for drug PEGylation are many: degree of PEGylation, determination of an ideal position within the molecule for PEG-linkage, evaluation of an ideal PEG type and PEG length, choice of PEGylation chemistry, preservation of the drug´s affinity, control of loss in specificity and selectivity, and the avoidance of the gain of unwanted binding effects.
Altogether, an extensive chemical and biological analysis of the PEGylated drug cannot be
avoided. In the context of hydrophobic ligands on the surface of PEG-coated nanoparticles,
the existence of individual PEG and ligand configurations comes into question, depending
on the PEG density and ligand properties. This aspect is important since the behavior of the
ligand in proximity to a dense PEG shell significantly influences the characteristics of the
particle in respect to its charge and its impact on cell binding and uptake. Two possible
configurations are known for PEG: brush and mushroom, differentiated by PEG chain
density. As a high PEG density is expected to yield a brush conformation with good shielding
properties and high particle mobility, this conformation is favored for PEG in nanoparticle
targeting in respect to ligand coupling. However, a high PEG density implies the risk for
hydrophobic ligand shielding, which could drastically minimize the exposure of the ligands
to the outer nanoparticle surface and therefore reduce the potential for multivalent receptor
binding.
Mechanistic concept of enzymatic nanoparticle activation in the context of diabetic nephropathy and rheumatoid arthritis
The concept of enzyme-based nanoparticle activation at the site of the disease as a further strategy for active targeting is discussed in the literature. In particular, metalloproteinases, which are expressed ubiquitously, are the focus of nanoparticle shell conversion to make underlying ligands available [55]. But no concept currently focuses on the induction of multivalency by enzyme driven ligand conversion on the surface of nanoparticles.
For earlier disease stages in RA, David A. Walsh and colleagues found that increased levels
of angiotensin-converting enzyme (ACE), a membrane metallopeptidase are present in the
synovial fluid of patients because of the increasing density of ACE-bearing stromal cells and
not because of an increase in vascular delivered ACE [29, 56]. Angiotensin-converting
enzyme primarily converts angiotensin I to the vasoactive angiotensin II and can therefore
be one reason for the progress of disease with respect to the formation of new small blood
vessels in the synovium [29]. Especially in the acute inflammation phase of arthritis,
bradykinin, substance P, CGRP, angiotensin II, prostaglandin E2, nitric oxide, histamine,
and hyaluronic acid (low molecular weight) contribute to angiogenesis. In the chronic phase,
other influences like vascular endothelial growth factor and other growth factors, such as
interleukins, TNF-α, TGF-β, and the platelet-derived endothelial cell growth factor play a
role. All these factors can be designated as angiogenic factors, and subclassified in acute and
chronic inflammation [29]. The inhibition of angiogenesis may be of therapeutic interest to
stop the progress of arthritic diseases. Angiotensin I-modified quantum dots may be useful
for further investigation of the influence of ACE in the progression of RA and for the
identification of target cells for enzymatically processed quantum dots. Moreover, these
quantum dots may be used as a diagnostic marker for different disease states. Since it is
known that, in addition to synovium derived cells, bovine chondrocytes in a 2D cell culture
express AT1 receptors for angiotensin II, cartilage cells could also be a potential target for
antiangiogenic drugs. Nanoparticles would reach these cells by simple diffusion, especially
in a pathological condition where the extracellular matrix is leaky because of cartilage
destruction. In preclinical research, such quantum dots could aid understanding of the
mechanism of action of ACE in RA after local administration into either inflamed or healthy joint space. In general, the selective inhibition of undesired angiogenesis is challenging because angiogenesis itself also has a physiological function and occurs in the absence of inflammation. Therefore, for the development of angiostatic therapeutics it is necessary to understand and distinguish among different regulatory mechanisms in both the healthy state and the disease state. In the healthy state, articular cartilage is avascular, and its nutrition is dependent from factors released from the synovium into the fluid-filled joint cavity. The normal synovium must be well supplied with capillaries to fulfill this function. This status is profoundly changed in the presence of inflammation because the morphology of the synovium is altered in respect to number, density, and spatial distribution of the vasculature [28]. In contrast to the healthy state, there is more vasculature in the disease state, but paradoxically the blood supply is inadequate. The consequences are hypoxia, hypercapnia, and acidosis [28]. If angiogenesis inhibitors reach off-targets, they could have potentially detrimental effects on wound healing or on the female reproductive capacity, for example [28]. Along with angiotensin I, substance P and bradykinin are also susceptible to ACEs. Compared to angiotensin, these proinflammatory kinins become inactive [57].
Substance P and bradykinin are known to induce plasma extravasation and to increase blood
flow [56, 58]. Therefore, it is therapeutically beneficial to stimulate ACE in the inflamed
joint, rather than to inhibit it.
References
[1] Pintér E, Pozsgai G, Hajna Z, Helyes Z, Szolcsányi J. Neuropeptide receptors as potential drug targets in the treatment of inflammatory conditions. Br J Clin Pharmacol 2014; 77: 5–20.
[2] Firestein GS, Kelley WN. Kelley's textbook of rheumatology: Neurologic Regulation of Inflammation. 9th ed. Philadelphia, PA [etc.]: Elsevier Saunders op. 2013 [i.e. 2012.
[3] Lai JP. Full-length and truncated neurokinin-1 receptor expression and function during monocyte/macrophage differentiation. Proceedings of the National Academy of Sciences 2006; 103: 7771–7776.
[4] Sabatino F, Di Zazzo A, Simone L de, Bonini S. The Intriguing Role of Neuropeptides at the Ocular Surface. The Ocular Surface 2017; 15: 2–14.
[5] Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol 2010; 6: 625–635.
[6] v. Euler, U. S., Gaddum JH. An unidentified depressor substance in certain tissue extracts.
The Journal of Physiology 1931; 72: 74–87.
[7] Dougados M. Synovial fluid cell analysis. Baillière's Clinical Rheumatology 1996; 10: 519–
534.
[8] Grässel S. The role of peripheral nerve fibers and their neurotransmitters in cartilage and bone physiology and pathophysiology. Arthritis Res Ther 2014; 16: 521.
[9] McDougall JJ. Arthritis and Pain. Neurogenic origin of joint pain. Arthritis Res Ther 2006; 8:
220.
[10] Hill R. NK1 (substance P) receptor antagonists--why are they not analgesic in humans?
Trends Pharmacol. Sci. 2000; 21: 244–246.
[11] Ricciotti E, FitzGerald GA. Prostaglandins and Inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology 2011; 31: 986–1000.
[12] Hong SK, Han JS, Min SS, et al. Local neurokinin-1 receptor in the knee joint contributes to the induction, but not maintenance, of arthritic pain in the rat. Neurosci. Lett. 2002; 322: 21–
24.
[13] Borbély É, Hajna Z, Sándor K, et al. Role of Tachykinin 1 and 4 Gene-Derived Neuropeptides and the Neurokinin 1 Receptor in Adjuvant-Induced Chronic Arthritis of the Mouse. PLoS ONE 2013; 8: e61684.
[14] Berger A, Paige CJ. Hemokinin-1 has Substance P-like function in U-251 MG astrocytoma cells: A pharmacological and functional study. Journal of Neuroimmunology 2005; 164: 48–
56.
[15] Lam FFY. Substance P and glutamate receptor antagonists improve the anti-arthritic actions of dexamethasone in rats. British Journal of Pharmacology 2010; 159: 958–969.
[16] Dickenson AH. Spinal cord pharmacology of pain. British Journal of Anaesthesia 1995; 75:
193–200.
[17] Johnson MB, Young AD, Marriott I. The Therapeutic Potential of Targeting Substance P/NK- 1R Interactions in Inflammatory CNS Disorders. Front. Cell. Neurosci. 2017; 10: 450.
[18] Richardson JD. Cellular Mechanisms of Neurogenic Inflammation. Journal of Pharmacology and Experimental Therapeutics 2002; 302: 839–845.
[19] Kramer MS, Cutler N, Feighner J, et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science (New York, N.Y.) 1998; 281: 1640–1645.
[20] Becher B, Prat A, Antel JP. Brain-immune connection: immuno-regulatory properties of CNS-resident cells. GLIA 2000; 29: 293–304.
[21] Banks WA, Kastin AJ. Blood to brain transport of interleukin links the immune and central nervous systems. Life Sciences 1991; 48: PL117-PL121.
[22] Dray A. Inflammatory mediators of pain. British Journal of Anaesthesia 1995; 75: 125–131.
[23] Muñoz M, Coveñas R. Involvement of substance P and the NK-1 receptor in human pathology. Amino Acids 2014; 46: 1727–1750.
[24] Schaible H-G, Banchet GS von, Boettger MK, et al. The role of proinflammatory cytokines in the generation and maintenance of joint pain. Annals of the New York Academy of Sciences 2010; 1193: 60–69.
[25] Garcia-Recio S, Gascón P. Biological and Pharmacological Aspects of the NK1-Receptor.
BioMed research international 2015; 2015: 495704.
[26] Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annual review of immunology 1996; 14: 397–440.
[27] Bonnet CS. Osteoarthritis, angiogenesis and inflammation. Rheumatology 2005; 44: 7–16.
[28] Stevens CR, Blake DR, Merry P, Revell PA, Levick JR. A comparative study by
morphometry of the microvasculature in normal and rheumatoid synovium. Arthritis and rheumatism 1991; 34: 1508–1513.
[29] Walsh D. Angiogenesis and arthritis. Rheumatology 1999; 38: 103–112.
[30] Millward-Sadler SJ, Mackenzie A, Wright MO, et al. Tachykinin expression in cartilage and function in human articular chondrocyte mechanotransduction. Arthritis & Rheumatism 2003;
48: 146–156.
[31] Vanderah T, Sandweiss A. The pharmacology of neurokinin receptors in addiction: prospects for therapy. SAR 2015: 93.
[32] Douglas SD. Neurokinin-1 receptor: functional significance in the immune system in reference to selected infections and inflammation. Annals of the New York Academy of Sciences 2011; 1217: 83–95.
[33] Fong TM. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Molecular Pharmacology 1992; 41: 24–30.
[34] DeFea KA, Vaughn ZD, O'Bryan EM, Nishijima D, Déry O, Bunnett NW. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin- dependent scaffolding complex. Proceedings of the National Academy of Sciences of the United States of America 2000; 97: 11086–11091.
[35] Caberlotto L, Hurd YL, Murdock P, et al. Neurokinin 1 receptor and relative abundance of the short and long isoforms in the human brain. Eur J Neurosci 2003; 17: 1736–1746.
[36] Patel HJ, Ramkissoon SH, Patel PS, Rameshwar P. Transformation of breast cells by truncated neurokinin-1 receptor is secondary to activation by preprotachykinin-A peptides.
Proceedings of the National Academy of Sciences 2005; 102: 17436–17441.
[37] Tuluc F, Lai JP, Kilpatrick LE, Evans DL, Douglas SD. Neurokinin 1 receptor isoforms and the control of innate immunity. Trends in Immunology 2009; 30: 271–276.
[38] Mammen M, Choi S-K, Whitesides GM. Polyvalent Interactions in Biological Systems:
Implications for Design and Use of Multivalent Ligands and Inhibitors. Angewandte Chemie International Edition 1998: 2754–2794.
[39] Hardman R. A Toxicologic Review of Quantum Dots: Toxicity Depends on Physicochemical and Environmental Factors. Environ Health Perspect 2006; 114: 165–172.
[40] Soo Choi H, Liu W, Misra P, et al. Renal clearance of quantum dots. Nat Biotechnol 2007;
25: 1165–1170.
[41] Bolonna AA. Partial agonism and schizophrenia. The British Journal of Psychiatry 2005; 186:
7–10.
[42] Gether U, Kobilka BK. G Protein-coupled Receptors. J. Biol. Chem. 1998; 273: 17979–
17982.
[43] Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends in Pharmacological Sciences 2007; 28: 397–406.
[44] Muñoz M. Involvement of substance P and the NK-1 receptor in pancreatic cancer. WJG 2014; 20: 2321.
[45] Seifert R, Wenzel-Seifert K, Gether U, Kobilka BK. Functional differences between full and partial agonists: evidence for ligand-specific receptor conformations. The Journal of
pharmacology and experimental therapeutics 2001; 297: 1218–1226.
[46] Bock A, Chirinda B, Krebs F, et al. Dynamic ligand binding dictates partial agonism at a G protein–coupled receptor. Nat Chem Biol 2013; 10: 18–20.
[47] Almeida TA, Rojo J, Nieto PM, et al. Tachykinins and tachykinin receptors: structure and activity relationships. Current medicinal chemistry 2004; 11: 2045–2081.
[48] Quartara L. The tachykinin NK1 receptor. Part I: Ligands and mechanisms of cellular activation. Neuropeptides 1997; 31: 537–563.
[49] Lee CM, Campbell NJ, Williams BJ, Iversen LL. Multiple tachykinin binding sites in peripheral tissues and in brain. European Journal of Pharmacology 1986; 130: 209–217.
[50] Gentilucci L, Marco R de, Cerisoli L. Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization.
Current pharmaceutical design 2010; 16: 3185–3203.
[51] Malavolta L, Cabral FR. Peptides: Important tools for the treatment of central nervous system disorders. Neuropeptides 2011; 45: 309–316.
[52] Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving
nanoparticle-based drug and gene delivery. Advanced Drug Delivery Reviews 2016; 99: 28–
51.
[53] Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv.
Drug Deliv. Rev. 2002; 54: 459–476.
[54] Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy 2008; 22: 315–329.
[55] Fay F, Hansen L, Hectors, Stefanie J. C. G., et al. Investigating the Cellular Specificity in Tumors of a Surface-Converting Nanoparticle by Multimodal Imaging. Bioconjugate Chem.
2017; 28: 1413–1421.
[56] Walsh DA. Angiotensin converting enzyme in human synovium: increased stromal
[125I]351A binding in rheumatoid arthritis. Annals of the Rheumatic Diseases 2000; 59: 125–
131.
[57] Emanueli C, Grady EF, Madeddu P, et al. Acute ACE inhibition causes plasma extravasation in mice that is mediated by bradykinin and substance P. Hypertension (Dallas, Tex. : 1979) 1998; 31: 1299–1304.
[58] Dray A, Bevan S. Inflammation and hyperalgesia: highlighting the team effort. Trends in Pharmacological Sciences 1993; 14: 287–290.
Chapter 2
Goals of the thesis
The goal of this thesis was to investigate the neurokinin-1 receptor as a potential target site for multivalent receptor blockade. Taking previous findings on multivalent G-protein- coupled receptor (GPCR) targeting into account, e.g. the increase in selectivity, specificity and ligand affinity to the target site, this work focused on challenging aspects of PEGylation of synthetic hydrophobic peptide and non-peptide NK
1R antagonists and their coupling to PEGylated quantum dots (QDs). Irrespective of neurokinin-1 receptor targeting, the principle of enzymatic peptide ligand activation on the surface of nanoparticles was investigated, based on angiotensin I to angiotensin II conversion by ACE and AT
1receptor targeting as a new tool for site-specific targeting.
As the neurokinin-1 receptor is involved in inflammation and pain, its role is also discussed in the context of autoimmune diseases, like RA, but also in cancer. An efficient antagonistic targeting could be highly beneficial in their therapy. Since the NK
1R is peripherally expressed on different cell types of the human body and its expression increases at the time when an immune response occurs, this receptor seems to offer great potential for selective and specific antagonistic surface receptor targeting with a minimizing effect of inflammation and pain. The interrelation of the neurokinin-1 receptor to inflammation and pain in general and how different ligands can interact with the neurokinin-1 receptor, as well as the cost- benefit of PEGylation of such ligands are described in Chapter 1.
Based on this scientific background, the expression status of the NK
1R was checked on different cell types, e.g. bovine chondrocytes as a potential target cell in RA, different cancer cell types, and transfected CHO cells as a positive control. Within these expression studies, the influence of the main neurokinin-1 receptor agonist, substance P was tested in respect to intracellular pathways. These studies are shown in Chapter 3.
The idea behind multivalent binding antagonists is to make potent ligands more selective
against cells with overexpression of the respective target receptor and the receptor blockade
itself more efficient because of a prolonged arrest of the multivalent binding ligand at the
target site. Fluorescent nanoparticles are a common tool to investigate such multivalent
interactions. Therefore, fluorescent PEGylated QDs were used for ligand coupling to the
surface of nanoparticles and to study their interactions with neurokinin-1 receptor positive
cells. The synthesis strategy and the nanoparticle characterization are shown in Chapter 4.
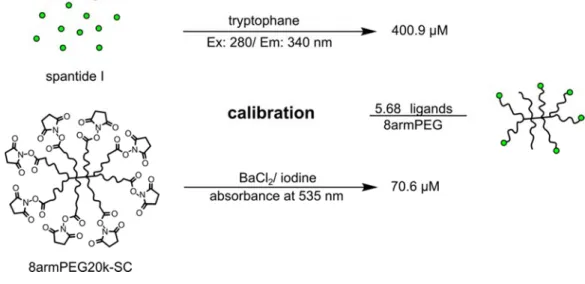
Besides PEGylated QDs, branched 8-arm PEGs were used for multivalent cell interaction studies. In contrast to QDs, PEGs are classified as non-toxic biomaterials and they do not interfere with luminescence-based calcium assays. Therefore, they offered an effective tool for determining their binding affinities as well as the ligand coupling efficiency. These binding studies are depicted in Chapter 5.
Besides peptide-based antagonists, a small molecular weight antagonist, named aprepitant was modified to make it amenable for further PEG coupling, either to branched PEGs or PEG-coated nanoparticles. The synthesis strategy is shown in Chapter 6.
Another new strategy for site-specific multivalent nanoparticle targeting is presented in
Chapter 7, the final chapter. This strategy is based on an enzyme driven activation
mechanism of ligands, which are immobilized on the surface of nanoparticles. For these
studies, the ACE driven angiotensin I to angiotensin II conversion was used and the
successful conversion was checked by AT
1receptor binding studies.
Chapter 3
Identification of target cell candidates with
neurokinin-1 receptor expression
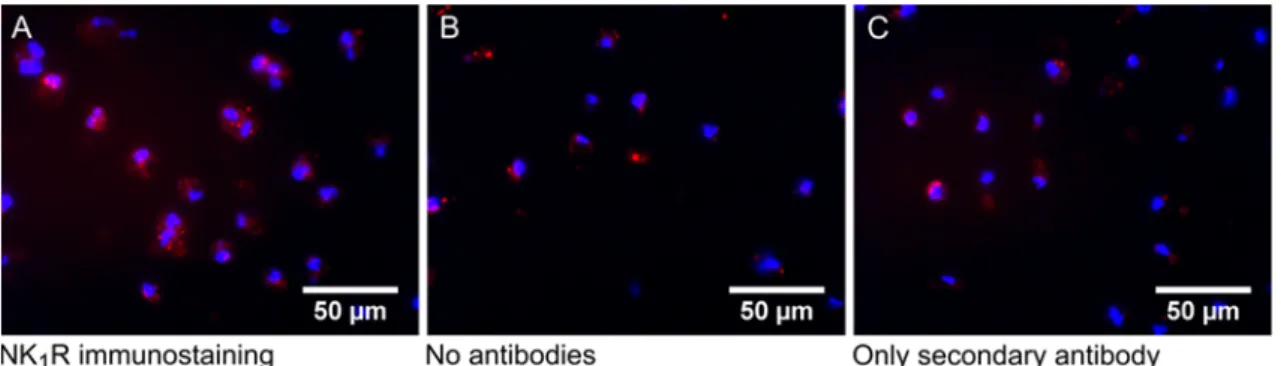
Abstract
The neurokinin-1 receptor (NK
1R) is a quite promising target receptor for multivalently binding nanoparticles for either tumor or non-tumor targeting because of its strict overexpression in pathological states and because of its action at the interface of the immune and nervous system. Though its direct role in the generation and perception of pain responses is controversial, its influence in the initiation and promotion of the immune response is certain. The presence of high local immune cell-derived cytokine concentrations leads to the release of substance P from peripheral afferent nerve terminals. Substance P is the major ligand for the NK
1R and triggers the immune response by pro-angiogenesis mechanisms, which facilitate the recruitment of further immune cells. This so-called neurogenic inflammation represents an effective way to protect the body from noxious stimuli. The main disadvantage of neurogenic inflammation is that there is a risk for irrepressible immune responses, especially in the case of cancer and autoimmune diseases (e.g. RA). Therefore, proinflammatory cytokine dependent NK
1R expression on various human glioblastoma and breast cancer cells and chondrocytes, obtained from bovine cartilage was investigated. Here, IL-1 dependent NK
1R mRNA expression was found in all tested cells. However, the existence of the functional full-length receptor version was not verified in calcium assays.
In bovine chondrocytes, substance P directly contributes to cartilage destruction by up-
regulation of the matrix-metalloproteinase MMP-13. Therefore, an effective antagonistic
treatment of the NK
1R may be a highly promising strategy to relieve typical inflammatory
symptoms in early RA and prevent further destructive processes in later disease stages.
Introduction
The peripheral nervous system and the immune system cannot be considered as two separately acting machineries [1]. The crosslink between both is drawn by the same molecular recognition pathways, which offer them a way to communicate. This mechanism is called neurogenic inflammation, with the goal of forming an integrated and rapid protective defense of the body. Due to the high speed of the signal transduction pathway of sensory and autonomic nerve fibers, which are widely distributed in peripheral tissues and organs, the neurogenic modulation of immunity is quite effective [1]. However, in the case of pathological states, as in cancer or autoimmune diseases (e.g. RA), this interaction also contributes to the exacerbation and progression of the disease [2].
The NK
1R and its primary agonist, substance P are two of the key players in neurogenic inflammation, since they are both upregulated during inflammation [3]. It is suggested that elevated substance P levels in plasma and synovial fluid may be a hallmark of chronic inflammation [4]. Substance P conveys vasodilatation, indicated by an increase in blood vessel size and an increase in permeability as well as neovascularization. Recent evidence suggests that substance P mediated stimulation of angiogenesis occurs in inflamed peripheral tissues like the synovial membrane in RA or different types of tumors [5,6]. Through these newly formed highly permeable blood vessels, immune cells are able to infiltrate into the joint cavity or into tumor tissues to release in turn cytokines like IL-1α and IL-1β and other pro-inflammatory factors, like TNF-α [7]. In the case of RA, the synovial membrane becomes hypertrophic and the high doses of inflammatory factors provoke the release of neuropeptides like substance P from articular afferent unmyelinated C nerve fibers within the soft tissues of the joint [7–9]. An additional factor for substance P release is local mechanical overload, which causes increasing compressive forces and hypoxia and may further sensitize these nerves and contribute to pain [10]. Prolonged peripheral nerve sensitization leads to enhanced responses of the spinal cord and thalamic neurons [11]
(Figure 1). The released substance P activates NK
1Rs, which are typically over-expressed
on glial cells of the CNS and peripheral infiltrated immune cells in the synovium and
synovial endothelial cells and even on articular chondrocytes [12–15]. In RA, a chronic stage
is reached, when the inflammatory symptoms last more than three months. In this state, the
ECM degrading matrix-metalloproteinases (MMPs), which are promoted by inflammatory transmitters and substance P [13].
Figure 1: The dominating part in neuronal innervations of an inflamed tissue is taken by sensory nerve fibers. In response to cytokine stimulation, the ends of these nerve fibers release various neuropeptides, including substance P, which cause vasodilatation and the recruitment of immune cells into the inflamed tissue. Reprinted from Nat Rev Rheumatol 9, 2, 117–126 by G. Pongratz and R. H. Straub, 2013. Copyright by Nature Publishing Group. Reprinted by permission [16].
An effective antagonistic targeting of substance P by a neurokinin-1 receptor blockade seems
to be a promising strategy to mitigate typical inflammatory and painful symptoms in either
cancer or in early and late stages of RA. For this reason, the NK
1R expression on different
tumor cell lines as well on bovine chondrocytes as potential target cells for multivalent
nanoparticle-based antagonistic NK
1R treatment was investigated. In this context, it was
additionally looked on NK
1R mRNA expression levels in response to stimulation with the
proinflammatory factor IL-1β. Further, whether substance P has an influence on MMP-1 and
MMP-13 expression on IL-1β stimulated bovine chondrocytes was investigated. The aspect
of ECM degradation in response to cytokines and neuropeptides is intriguing since it is a
substantial requirement for nanoparticle diffusion into cartilage and subsequent NK
1R
targeting on chondrocytes. Because NK
1R mRNA levels do not directly reflect the functional
receptor density on the cell surface, calcium assays were performed with all cell lines to
identify a target cell with high NK
1R density for multivalent drug testing.
Materials and methods
All chemicals were obtained from Sigma Aldrich (Taufkirchen, Germany) in analytical grade unless stated otherwise. Dulbecco’s phosphate-buffered saline (DPBS) pH 7.4 consisting of 1.5 mM KH
2PO
4, 8 mM Na
2HPO
4, 2.7 mM KCl and 138 mM NaCl was purchased from Life Technologies/Thermo Scientific (Darmstadt, Germany). Ultrapure water was obtained from a Milli-Q water purification system (Millipore, Billerica, MA, USA).
Tumor cell lines, U87 MG gliloblastoma, MDA-MB-231 and MCF-7 breast cancer cell lines were purchased from ATCC [17,18]. U87 MG cells were grown in Eagle´s Minimum Essential Medium (EMEM) without pyruvat with 10% FCS, MDA-MB-231 in McCoy´s medium with 5% FCS and MCF-7 in EMEM with pyruvat and 10% FCS. Primary bovine chondrocytes were isolated by Johanna Lempp from the department of Pharmaceutical Technology (Universität Regensburg) from freshly obtained bovine knee joints and grown in cell culture medium (CCM), consisting of DMEM (Gibco/Thermo Scientific, Darmstadt, Germany), 10 mM HEPES, non-essential amino acid (Gibco/Thermo Scientific, Darmstadt, Germany), 46 µg/ml proline, 50 µg/ml ascorbic acid, 1% Penicillin/Streptomycin (Gibco/Thermo Scientific, Darmstadt, Germany), 10% FCS. DBA-1J mouse knee joints were kindly provided from Prof. Rainer Straub (Klinik und Poliklinik für Innere Medizin, Universität Regensburg). Dr. Ralf Schwandner (Amgen Research GmbH, BioPark Regensburg) kindly provide a stable transfected neurokinin-1 receptor expressing CHO cell line. Cells were grown in DMEM:F12 (1:1) medium with 1.2 g/l NaHCO
3, 15 mM HEPES, 1% Penicillin-Streptomycin, 10% FCS and 600 µg/ml hygromycin B (Carl Roth, Karlsruhe, Germany) supplementation.
The peqGOLD Total RNA Kit and the peqGOLD DNase I Digest Kit were used to extract
mRNA and to remove potentially present DNA. For reverse transcription, two kits were
used: The AffinityScript QPCR cDNA Synthesis Kit was bought from Agilent Technologies,
Waldbronn Germany and the PeqGOLD cDNA Synthesis Kit H Minus was bought from
Peqlab, Erlangen, Germany. For classical Polymerase chain reaction (PCR), the peqGOLD
Taq-DNA-Polymerase Kit from Peqlab, Erlangen, Germany was used. As reaction buffer,
the 10x reaction buffer S was always chosen, with a MgCl
2content of 15 mM. For
quantitative real-time PCR, the SYBR Green qPCR Kit was obtained from Agilent Technologies, Waldbronn, Germany.
Human interleukin-1α (IL-1α) was purchased from PeproTech, Hamburg, Germany. Human interleukin-1β (IL-1β) was purchased either from MACS Miltenyi Biotec, Bergisch Gladbach, Germany or from PeproTech, Hamburg, Germany. Reconstitutions were made by dissolving 10 µg of provided interleukins in 100 µl sterile filtered Millipore water to obtain 0.1 mg/ml stock solutions. Working solutions of 0.01 mg/ml were prepared in 0.1% BSA in DPBS.
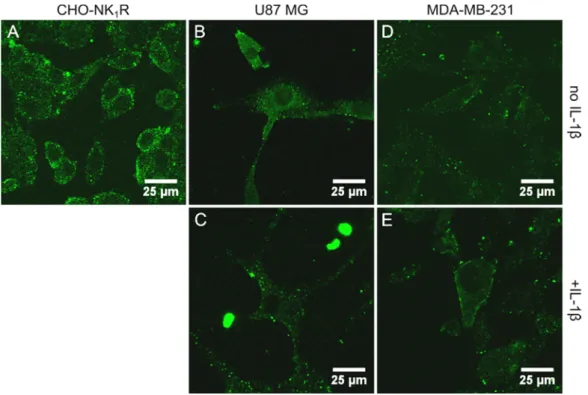
Neurokinin-1 receptor expression studies in 2D cancer cell culture upon 24 hours interleukin-1β stimulation by RT-PCR
U87 MG and MDA-MB-231 cells were seeded in a six-well cell culture plate in a cell density of 0.5*10
6cells per well and were grown overnight at 37°C and 5% CO
2in a medium with 10% FCS. Stimulation over 24 hours was started 12 hours after cell adherence in a medium without FCS. On the second day after seeding, stimulation time points of 6 hours, 3 hours, 1 hour and the control without stimulation were prepared equivalently.
After IL-1β stimulation, the medium was removed and replaced by 1 ml phenol containing
TRIzol reagent for cell lysis and incubated for 5 min at room temperature. In each well, the
total cell lysate was removed with the help of a cell scraper and transferred to a 2 ml
Eppendorf tube. For phase separation, 200 µl of chloroform were added and mixed by
thorough up and down pipetting for 15 seconds. To induce complete phase separation, the
mixture was centrifuged at 12000 rcf for 15 min at 4°C. The upper phase was transferred in
a fresh Eppendorf tube and washed with 500 µl isopropanol by gentle shaking, and RNA
was precipitated by incubation at room temperature for 10 min. The extracted RNA was
sedimented by a further centrifugation at 14000 rcf for 30 min at 4°C. Then the supernatant
was removed and the obtained RNA pellet was air dried. Then the pellet was washed in 1 ml
75% ethanol by gentle shaking and a centrifugation step at 9400 rcf for 5 min at 4°C. The
RNA pellet was dried again after removal of the supernatant. The obtained RNA was
dissolved in 15 µl RNase free water and heated to 55°C for at least 10 min. The obtained
samples were stored at -80°C until use. The RNA content was determined with a NanoDrop UV meter. For cDNA generation, 2 µg RNA were reverse-transcribed with an oligo(dT) primer and random primer mix, consisting of 170 ng and 30 ng respectively. It was considered that an existing short receptor version, lacking some C-terminal amino acids for G-protein binding, has no Poly-A tail for oligo(dT) primers. With the obvious excess of oligo(dT) primers, the mRNA of full-length receptor version should be preferably transcribed in the RT reaction. The resulting cDNAs were used as templates for amplification in PCRs.
Neurokinin-1 receptor expression studies in 2D bovine chondrocyte cell culture by 24 hours IL-1α and IL-1β stimulation by RT-PCR
To evaluate whether the NK
1R is expressed in general and whether there is a change in expression in the state of inflammation, primary bovine chondrocytes were stimulated in a 2D culture with two different interleukins, and mRNA was subsequently isolated for RT-PCR. Therefore, bovine chondrocytes of the first passage were cultured in T25 cell culture flasks (Corning, Wiesbaden, Germany) in CCM. Confluent cells were washed with warm DPBS and then stimulated with interleukins, either 10 ng/ml IL-1α or IL-β (PreproTech, Hamburg, Germany) in phenol red free CCM without FCS and 0.1% BSA and incubated for 24 h at 37°C and 5% CO
2atmosphere. The peqGOLD Total RNA Kit and the peqGOLD DNase I Digest Kit were used to extract mRNA and to remove potentially present DNA. Both kits were used according to the manufacturer´s instructions. The RNA content was determined at a NanoDrop UV meter. Then 1 µg RNA was reverse-transcribed with the peqGOLD cDNA Synthesis Kit H Minus, using an oligo(dT) primer and random primer mix of 170 ng to 30 ng. The resulting cDNAs were used as templates for amplification in PCRs.
Polymerase chain reaction (PCR)
There are two versions of neurokinin-1 receptors - a full-length form and a truncated form
generated by alternative splicing [19]. Compared to the full-length receptor version, the
truncated receptor lacks 96 amino acids in the C-terminal part of the receptor sequence in
the cytosol and is therefore not able to bind and activate a G-protein and trigger intracellular calcium release upon extracellular agonist binding [20,21]. For the evaluation of cell lines, which express both types of neurokinin-1 receptors, appropriate primer pairs for PCR that span the cDNA for the full-length and the truncated receptor version were used. The PCR primers were ordered from Eurofins Genomics, Ebersberg, Germany and are listed in Table 1.
Table 1: Primer sequences
Target Sequence (5´-> 3´) Tm in °C GC content in %
Human NK1R [22] Forw.: AGGACAGTGACGAACTATTTTCTGG
Rev.: CTGCTGGATAAACTTCTTCAGGTAG 61.3
61.3 44.0 44.0
Bovine NK1R [23] Forw.: TATTTACTCCATGACGGCTG
Rev.: AGTCCCCAGGGATCTCACT 55.3
58.8 45.0 57.9
Human GAPDH Forw.: CTGACTTCAACAGCGACACC
Rev.: CCCTGTTGCTGTAGCCAAAT 59.4
57.3 55.0 50.0
For the detection of human neurokinin-1 receptor expression levels, a PCR was performed using the peqGOLD Taq-DNA-Polymerase Kit. For each reaction, a total volume of 25 µl with a primer content of 20 pmol (human NK
1R) and 10 pmol (bovine NK
1R) and 1 µl of cDNA template were used [22]. As reaction buffer, 10x reaction buffer S was chosen with a MgCl
2content of 15 mM. All other components were applied according the manufacturer´s instructions.
The PCR program was based on those mentioned in the literature [22]:
1. Denaturation 94°C 9 min
1)/2 min
2)/3)2. Denaturation 94°C 30 sec
1)3)/10 sec
2)3. Annealing 50°C
1)/55°C
2)/3)30 sec 45
1)/40
2)/35
3)cycles 4. Elongation 72°C 30 sec
1)/35 sec
2)/15 sec
3)5. Final elongation 72°C
1)/2)/76°C
3)7 min
1)/2 min
2)/6 min
3)6. 4°C ∞
1)
human NK
1R,
2)bovine NK
1R,
3)bovine MMP-1 and MMP-13 (see following section)
The amplified cDNA was finally observed in a gel electrophoresis with 2% agarose (Invitrogen/Thermo Scientific, Darmstadt, Germany) in 1x TAE buffer. The bands were detected with 0.4 µg/ml ethidium bromide, which was added to the pre-solid gel. For sample load, 20 µl of PCR reaction product were mixed with 4 µl 6x Loading Dye (Peqlab, Erlangen, Germany). For fragment size determination either the peqGOLD 50 bp (Peqlab, Erlangen, Germany) or 100 bp DNA ladder (Invitrogen/ ThermoFisher Scientific, Darmstadt, Germany) were used.
RT-PCR and qPCR for MMP-1 and MMP-13 mRNA
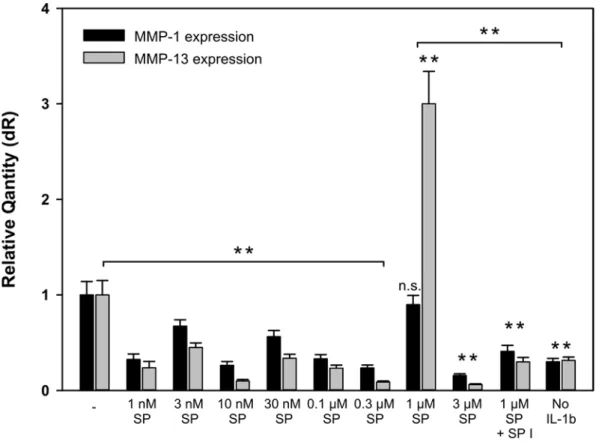
expression analysis upon substance P stimulation in 2D bovine chondrocyte cell culture
Because of the lack of specific bovine neuokinin-1 receptor primers to study the influence of substance P on receptor expression in real-time qPCR, substance P mediated matrix- metalloproteinase (MMP-1 and MMP-13) expression was investigated, which would be an indirect proof for the existence of neurokinin-1 receptors on the surface of chondrocytes.
Therefore, two different stimulation experiments were performed. In the first experiment,
various substance P concentrations in a fixed timeframe of 24 hours were tested, starting
from 1 nM and increasing up to 3 µM substance P. In the second experiment, a fixed
concentration of 1 µM substance P was used, and the stimulation time was varied from
1 hour up to 48 hours. The RNA extraction and reverse transcription were performed as
described in the former section. The resulting cDNAs were used as templates for
amplification in polymerase chain reactions and for quantitative real-time PCR (qPCR)
analysis, where expression levels were normalized to GAPDH housekeeping gene
expression. Template cDNA was used in a volume of 10.5 µl and mixed to a final volume
of 25 µl with components of the SYBR Green qPCR Kit (Agilent Technologies, Waldbronn,
Germany), according to the manufacturer´s instructions, and mixed with 10 pmol of
prepared bovine MMP-1, bovine MMP-13, or bovine GAPDH primer mix (primer
sequences and characteristics are shown in Table 2). Real-time gene amplification was
performed in a 96-well non-skirted plate with adhesive foil seal (4titude®Ltd., Berlin,
Germany), using a spectrofluorometric thermal Mx3005P QPCR System (Agilent
Technologies, Santa Clara, CA, USA). The cycling program started with an initial cycle at
95°C for 15 minutes, followed by 40 cycles, each starting at 95°C for 10 seconds and decreasing the temperature to 60°C and holding for 30 seconds, and ended with a final cycle at 95°C for 1 min, decreasing to 55°C and holding for 30 seconds, then increasing again to 95°C for 30 seconds. Data evaluation was performed with the system connected QPCR software MxPro (Agilent Technologies, Waldbronn, Germany).
Table 2: Primer sequences
Target Sequence (5´-> 3´) Tm in °C GC content in %
Bovine MMP-1 [24] Forw.: GCTTATGAGGTTGCCGACAG Rev.: ACGTTTTCCCAGTATCTTCCTC
59.4 58.4
55.0 45.5 Bovine MMP-13 [24] Forw.: CGCGGAGAAACACTGATCTT
Rev.: TCATAGGCGGCATCAATACG
57.3 57.3
50.0 50.0 Bovine GAPDH Forw.: TTCTGGCAAAGTGGACATCG
Rev.: CGTTCTCTGCCTTGACTGTG
57.3 59.4
50.0 55.0