Totalsynthese von Spirastrellolide F Methylester
281
0
0
Volltext
(2)(3)
(6)(7)
(8)(9)
(10)(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
(42)
(43)
(44)
(45)
(46)
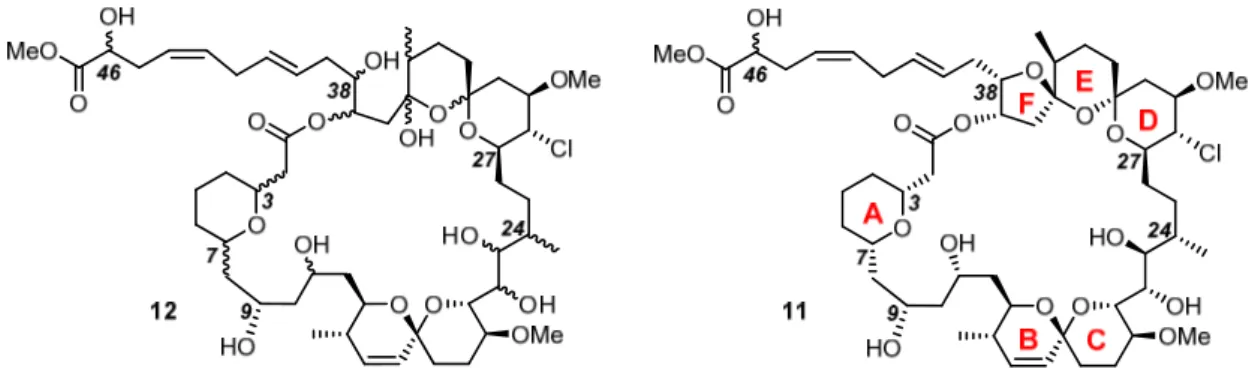
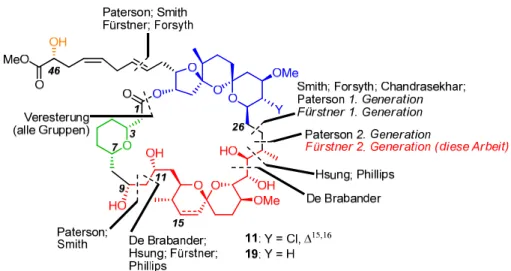
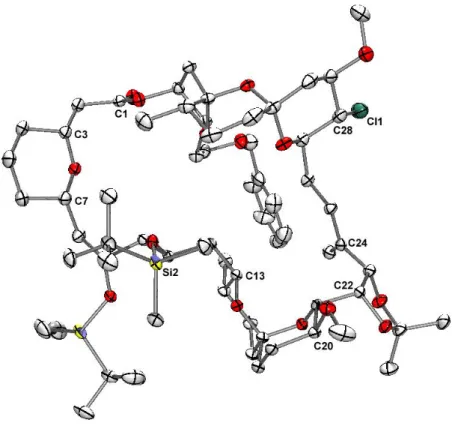
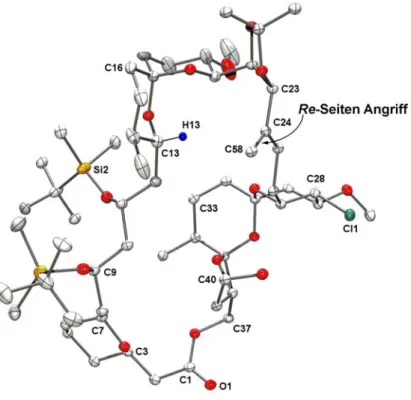
Abbildung


+7




ÄHNLICHE DOKUMENTE
Die vereinigten organischen Phasen werden mit gesättigter wäßriger NaHCO 3 -Lösung (2 ml) gewaschen, getrocknet (MgSO 4 ), filtriert und im Vakuum konzentriert. Nach 1h werden
Anschließend wurde die wässrige Phase mit Et 2 O (4x60 mL) extrahiert, die vereinigten organischen Phasen über MgSO 4 getrocknet und das Lösemittel unter vermindertem Druck
Die wässrige Phase wird viermal mit einer Mischung aus 15 % Ethylacetat in Petrolether extrahiert, die vereinigten organischen Phasen werden über Magnesiumsulfat getrocknet,
Die wässrige Phase wurde noch 2 x mit Dichlormethan extrahiert, die vereinigten organischen Phasen über Natriumsulfat getrocknet, filtriert und im Vakuum vom Lösungsmittel
Die wässrige Phase wurde dreimal mit DCM extrahiert und die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und das Lösungsmittel im
Das Gemisch wurde mehrmals mit 10 mL Diethylether extrahiert und die vereinigten organischen Phasen über Na 2 SO 4
Die wässrige Phase wurde dreimal mit Ethylacetat extrahiert, die vereinigten organischen Phasen wurden über Na 2 SO 4 getrocknet, filtriert und das Lösungsmittel
Nach der Phasentrennung wird die wäßrige Phase mehrfach mit Diethylether extrahiert, die vereinigten organischen Phasen mit Na 2 SO 4 getrocknet und Lösungsmittel
