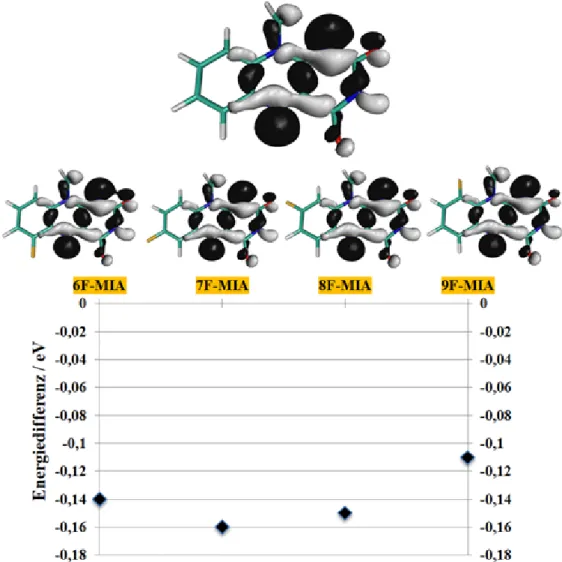
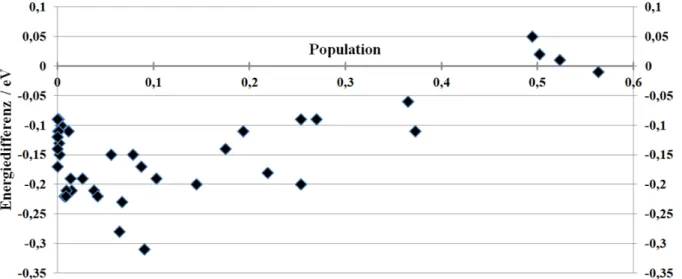
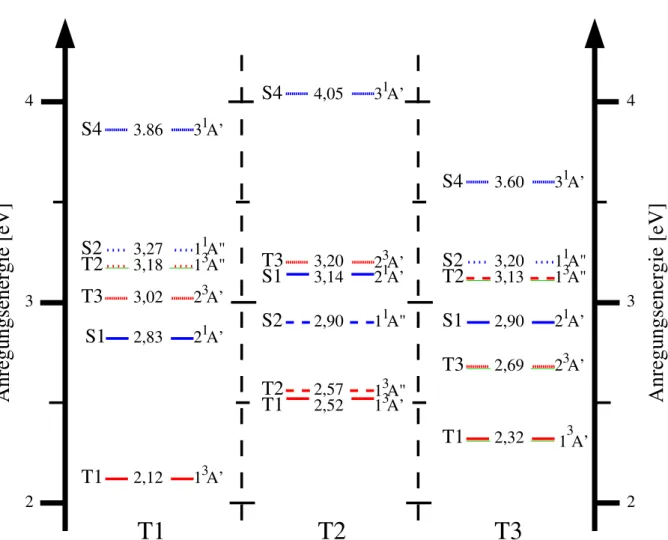
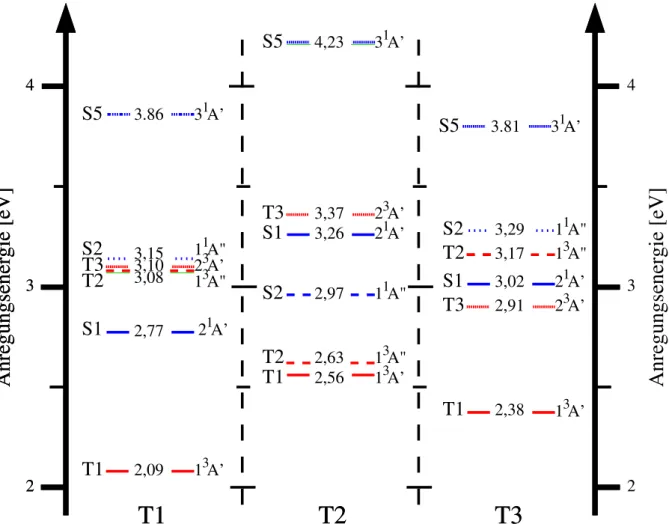
Photophysik fluorierter Flavine in Lösung aus quantenchemischer Sicht
Institut für Theoretische Chemie und Computerchemie der Heinrich-Heine-Universität
Düsseldorf
Abschlussarbeit
zur Erlangung des akademischen Grades Master of Science
vorgelegt von
Mario Bracker
geboren am 02.02.1993 in Haan
im Oktober 2017
Eidesstattliche Erklärung
Hiermit versichere ich, die vorliegende Masterarbeit ohne Hilfe Dritter und nur unter Verwen- dung der von mir angegebenen Quellen sowie Hilfsmittel verfasst zu haben.
Wörtlich oder sinngemäß übernommene Stellen wurden in den Quellen kenntlich gemacht, so- wie Hilfsmittel angegeben.
Die Arbeit hat in dieser oder vergleichbarer Form noch keinem anderem Prüfungsgremium vorgelegen.
Datum: Unterschrift:
Danksagungen
An dieser Stelle möchte ich mich recht herzlich bei allen Personen bedanken, die zum Gelingen dieser Arbeit beigetragen haben. Allen voran gilt meine Dankbarkeit Frau Professorin Doktorin Christel M. Marian, die mich mit diesem interessanten Forschungsthema betraut hat und sich großzügig Zeit für mich genommen hat.
Ein großer Dank geht auch an Doktor Martin Kleinschmidt, der stets die passenden Antworten auf meine Fragen parat hatte und mich beim Korrekturlesen maßgeblich unterstützt hat.
Vielen Dank auch an Privatdozent Doktor Oliver Weingart, zum einen für die Beteiligung am Korrektur lesen, zum anderen für die Einweihung in die interessante Methodik der Quan- tenmechanik/Molekülmechanik. Bei aufkommenden Fragen und vor allem Problemen konnte er immer Abhilfe schaffen.
Ebenfalls bedanken möchte ich mich bei Professor Doktor Gilch für die Übernahme des Zweit- gutachtens.
Im Allgemeinen möchte ich mich beim gesamten Arbeitskreis für die Offenheit und Hilfsbe- reitschaft bedanken. Hervorzuheben ist hierbei Fabian Dinkelbach, der sich das ein oder andere mal mit meinen Fragen und Problemen beschäftigt hat.
Ich freue mich auf die weitere Zusammenarbeit.
Inhaltsangabe
Diese Arbeit thematisiert die Photophysik der Flavine. Das Hauptaugenmerk liegt dabei auf dem Einfluss der Fluorierung am annelierten Benzolring auf die Photophysik des Chromophors.
Konkret wird im Rahmen dieser Arbeit der Relaxationsprozess von Methylisoalloxazin (MIA) sowie der Derivate 6F-MIA, 7F-MIA, 8F-MIA und 9F-MIA im Zuge elektronischer Anregung nachvollzogen. Dies geschieht, indem für Geometrieoptima [(TD)DFT] relevanter Zustände (S0, S1,S2,T1,T2,T3) Anregungsenergien (DFT/MRCI) errechnet werden - in der Konsequenz kön- nen Relaxationskanäle, angeschlossen an Photoanregung, skizziert werden. Diesbezüglich wer- den Fluoreszenzraten und Spin-Bahn-Kopplungen errechnet. Die Auswirkung der Fluorierung in Abhängigkeit von der Position am Chromophor wird zum einen phänomenologisch betrach- tet und diskutiert, zum anderen werden die Effekte bezüglich der atomzentrierten Populationen einzelner Orbitale erläutert - der Einfluss auf konkrete Orbitalenergien wird nachvollzogen.
Diese Studie setzt sich zum einen mit den Eigenschaften der untersuchten Flavine in der Gas- phase auseinander, zum anderen werden Lösungsmittelmodelle für Wasser entworfen: Basie- rend auf impliziter und expliziter Methodik werden Modelle bezüglich ihrer Anwendung und Resultate verglichen. Der explizite Ansatz erfolgt mit Hilfe von QM/MM-Methodik (Quanten- mechanik/Molekülmechanik) und verfolgt das Ziel der präzisen Beschreibung von Wasserstoff- brückenbindungen.
Rechenergebnisse unter Verwendung der Solvatationsmodelle werden im Hinblick auf poten- tielle Anwendungen [Biodiagnostik, Photodynamische Therapie (PDT)] mit spektroskopischen Werten verglichen. Im Hinblick auf diesen Vergleich zeigt sich, dass keines der fluorierten De- rivate besonders geeignet für potentielle Anwendungen in der Biodiagnostik beziehungsweise Krebstherapie (PDT) ist; allerdings konnte der Einfluss der Fluorierung auf die Energetik und den Relaxationsprozess detailliert nachvollzogen werden, sodass weiterführende Studien mit anderen Fluorierungsmustern systematisch angegangen werden können.
Abstract
The central topic of this study is the photophysics of flavins - it is focussed upon the influence of fluorination of the annealed benzene ring on the photophysics of the cromophore. In concrete terms this examination deals with the relaxation process of Methylisoalloxazine (MIA) and its derivatives 6F-MIA, 7F-MIA, 8F-MIA und 9F-MIA in the course of photoexcitation. Concer- ning this complex matter, geometries of selected electronic states are optimized [(TD)DFT]
(S0,S1, S2, T1, T2, T3) and excitation energies are calculated (DFT/MRCI). Subsequently the relaxation paths are outlined - fluorescence rates and spin-orbit-couplings are computed. On the one hand the impact of fluorination on the relaxation process is evaluated phenomenologically, on the other hand the influence concerning the energies of selected orbitals is examined as well as explained.
This study is divided into an analysis of the photophysics of the investigated flavins in ga- seous phase, hereupon the changes in aqueous phase are determined. With regard to the latter case, solvation models based on implicit and explicit methods are delineated and compared re- ferring to the results and application. The explicit approach is realized by means of QM/MM- methodology (Quantum/Molecular mechanics), aiming towards a precise description of the covalent character of hydrogen bonds.
With regard to potential applications of the investigated flavins [Biodiagnostics, Photodynamic therapy (PDT)], the results using the designed solvation models are in consequence compared to spectroscopic data. It arises that none of the fluorinated derivatives is particularly qualified
Inhaltsverzeichnis
1. Einleitung 1
1.1. Flavine in vivo, vitro - et silico . . . 2
1.2. Motivation . . . 6
I. Theorie & Praxis 9
2. Theoretischer Hintergrund 11 2.1. Motivation der Schrödingergleichung . . . 112.2. Das Abklingen angeregter Zustände . . . 14
2.2.1. Helle und dunkle Zustände . . . 17
2.3. Dichtefunktionaltheorie (DFT) . . . 18
2.3.1. Zeitabhängige Dichtefunktionaltheorie (TD-DFT) . . . 21
2.4. Elektronenkorrelation . . . 24
2.4.1. DFT/MRCI . . . 27
2.5. Solvatationsmodelle . . . 28
2.5.1. Conductor-like screening model (COSMO) . . . 30
2.5.2. Quantenmechanik/Molekülmechanik (QM/MM) . . . 31
2.5.2.1. Mechanische & Elektrostatische Einbettung . . . 32
2.5.2.2. Velocity-Verlet-Algorithmus . . . 34
2.6. Spin-Bahn-Kopplung . . . 35
3. Technische Details 39 3.1. Software . . . 39
3.2. Basissatz . . . 39
3.3. Funktional . . . 39
3.4. Geometrieoptimierung . . . 40
3.5. DFT/MRCI . . . 40
3.6. Solvatationsmodelle . . . 40
3.6.1. COSMO . . . 40
3.6.2. Molekülmechanik . . . 40
3.6.3. Modellierung . . . 41
3.6.3.1. Modell I . . . 41
3.6.3.2. Modell II . . . 41
Inhaltsverzeichnis
II. Auswertung & Interpretation 43
4. Resultate 45
4.1. Struktur der Geometrie . . . 45
4.2. Elektronische Struktur . . . 48
4.2.1. Helle und dunkle Zustände . . . 49
4.2.2. Relaxationspfad . . . 55
4.2.3. Einfluss der Fluorierung . . . 56
4.2.3.1. Vergleich ausgewählter Orbitale . . . 57
4.3. Fluorierte Derivate . . . 65
4.3.1. 6F-Methylisoalloxazin (6F-MIA) . . . 66
4.3.2. 7F-Methylisoalloxazin (7F-MIA) . . . 68
4.3.3. 8F-Methylisoalloxazin (8F-MIA) . . . 70
4.3.4. 9F-Methylisoalloxazin (9F-MIA) . . . 72
4.3.5. Vergleich . . . 74
4.3.5.1. S0-Geometrie . . . 74
4.3.5.2. S1-Geometrie . . . 76
4.3.5.3. S2-Geometrie . . . 78
4.3.5.4. T1-Geometrie . . . 80
4.3.5.5. T2-Geometrie . . . 82
4.3.5.6. T3-Geometrie . . . 84
4.4. Einfluss der Solvatation auf die Zustände . . . 86
4.4.1. Anregungsenergien in Wasser . . . 92
4.4.1.1. MIA - H2O . . . 93
4.4.1.2. 6F-MIA - H2O . . . 97
4.4.1.3. 7F-MIA - H2O . . . 101
4.4.1.4. 8F-MIA - H2O . . . 105
4.4.1.5. 9F-MIA - H2O . . . 109
4.5. Fluoreszenz . . . 113
4.6. Interkombination . . . 115
5. Diskurs 117 5.1. Solvatationsmodelle . . . 117
5.2. Vergleich mit Messwerten . . . 121
6. Résumé 127 6.1. Ausblick . . . 128
III. Anhang i
7. Molekülorbitale iii
8. Stabilisierungsenergien von Grenzorbitalen ix
8
9. Konstitutionen der Anregungen xvii
10.Populationsanalyse xxv
11.Konstitutionen der Anregungen in Wasser - Modell III xxxiii 11.1. Sampling-Energien (Modell III) . . . xliv 12.Anregungskonstitutionen in Wasser - Modelle IV-V li
Abkürzungsverzeichnis
AMFI AtomicMean Field Integral AO AtomOrbital
ATP AdenosinTriPhosphat
BHLYP Becke Half and Half Lee-Yang-Parr BLUF Blue-Light sensorsUsing FAD
B3LYP Becke-3-Lee-Yang-Parr
CC2 Linear response approximate CoupledCluster methods CI Conical Intersection
COBRAMM Computations OBtained by Running Ab-initio and MolecularMechanics COSMO COnductor-like Screening MOdel
cry Cryptochrom
CSF Configuration State Function CT Charge Transfer
CTP CytidinTriPhosphat FAD Flavin Adenin Dinukleotid
FbFP FMN-bindendes FluoreszenzProtein FCKW Fluor-Chlor-KohlenWasserstoffe FMN FlavinMonoNukleotid
GAFF Generalized Atomic Force Field GFP Green Fluorescent Protein
HF Hartree-Fock
HOMO Highest OccupiedMolecular Orbital IC Internal Conversion
IR Infrared Emission ISC InterSystem Crossing
IVR Internal Vibrational Redistribution LCAO Linear Combination of Atom Orbitals LE Local Excitation
LOV Light-Oxygen-Voltage-sensing
LUMO Lowest Unoccupied MolecularOrbital
(MC)SCF (Multi-ConfigurationSelf-Consistent-Field MIA MethylIsoAlloxazin
MRCI Multi-Reference ConfigurationInteraction
MRPT2 Second-orderMulti-Reference Perturbation Theory KS Kohn-Sham
phot Phototropin
QM/MM QuantenMechanik/MolekülMechanik
(TD)DFT (Time-Dependant) Density Functional Theory PFT PerFluorierte Tenside
PTFE PolyTetraFluorEthen SD Singles and Doubles SOC Spin-Orbit Coupling SOMF Spin-Orbit Mean Field SPOCK SPin-Orbit Coupling Kit
TIP3P three-site Transferable Intermolecular Potential TZVP Triple-Zeta Valence Polarisation
UDFT Unrestricted DFT VIS VISible
VR Vibrational Relaxation
WBB WasserstoffBrückenBindung xc Exchange-correlation
II
1. Einleitung
Das den Flavinen zugrundeliegende Strukturmotiv ist das Ringsystem des Isoalloxazin, eines Heterozyklus, der sich von Pteridin ableitet. Flavine unterscheiden sich von Isoalloxazinen durch die Methylgruppen an den Positionen 8 und 9, sowie einen organischen Rest am zentral gele- genen Stickstoff Nummer 10. Dieser Rest ist das Unterscheidungsmerkmal natürlich vorkom- mender Flavine, so kann es sich beispielsweise um eine Ribitylgruppe (Riboflavin1, VitaminB2, Lactoflavin; siehe Abbildung 2) handeln. Ausgehend vom Riboflavin ergibt die Phosphorylie- rung der endständigen Hydroxygruppe des Ribitylrests Flavinmononukleotid [FMN, Riboflavin- 5’-(dihydrogenphosphat); siehe Abbildung 2], ein Coenzym in diversen Oxidoreduktasen. Ein weiterer natürlicher Elektronenüberträger ist das Flavin-Adenin-Dinukleotid (FAD; siehe Ab- bildung 3); strukturell unterscheidet sich dieses von FMN durch eine weitere Phosphatgruppe, an welche Adenin, überbrückt durch Ribose (Nukleotid), gebunden ist.
Abbildung 1.: Abbildung des den Flavinen zugrundeliegenden Strukturmotivs. Der eigentliche Chromophor, das Isoalloxazin (IA), ergibt sich mit R = H. Flavine sind durch Methylgruppen für R0 und R00, sowie einen spezifischen Rest verknüpft mit N(10) gekennzeichnet.
17,8-Dimethyl-10-(D-ribo-2,3,4,5-tetrahydroxypentyl)-3H,10H-benzo[g]pteridin-2,4-dion (IUPAC)
KAPITEL 1. EINLEITUNG
1.1. Flavine in vivo, vitro - et silico
Die Stoffklasse der Flavine ist ein elementarer Baustein im Stoffwechsel von sowohl Prokaryoten als auch Eukaryoten. Bei Vitamin B22(Riboflavin), handelt es sich um das zuerst entdeckte Fla- vin. Die Isolation erfolgte 1879 aus Kuhmilch [Bly79] (Lactochrom, Lactoflavin). Dieses Flavin ist Bestandteil unserer täglichen Nahrung, es kommt sowohl als ungebundenes Riboflavin als auch gebunden an Eiweiße in Milch(produkten), Gemüse (Broccoli, Spargel, Spinat), Vollkorn- produkten, Fisch, Muskelfleisch und Eiern vor; Mangel des essentiellen Riboflavins (Vitamin B2) verursacht Wachstumsstörungen, Hautkrankheiten und Haarausfall. Die Synthese erfolgt seit 1990 biotechnologisch im industriellen Maßstab: Der Wildtyp des Pilzes Ashbya gossypii produziert Riboflavin aus Biomasse, alternativ findet die Herstellung auch mit Hilfe gentech- nisch veränderter Stämme von Bacillus subtilis statt [Sah13]. Vitamin B2 ist für den Stoff- wechsel von fundamentaler Bedeutung, da es als Vorstufe für Koenzyme auf Flavinbasis (FMN, FAD) dient. Abgesehen von seiner Funktion in vivo wird Riboflavin als Lebensmittelfarbstoff3 (E101) eingesetzt. Ein weiteres Einsatzgebiet ist die Kontrolle von Reinigungsprozessen in der Pharmaindustrie, da es bereits in geringen Konzentrationen unter Beleuchtung mit UV-Licht erkenntlich ist. In Säugetieren erfolgt angeschlossen an die Aufnahme des Vitamins über die Nahrungskette unter anderem die Umwandlung in Flavinmononukleotid mittels Flavokinase.
Abbildung 2.: Abbildung der Strukturmotive von Riboflavin (Vitamin B2, Lactoflavin) und dem daraus hervorgehenden Flavinmononukleotid (FMN).
Das Mononukleotid fungiert als Coenzym in diversen Oxidoreduktasen [(zum Beispiel NADH- Dehydrogenase (Komplex I der mitochondrialen Atmungeskette)]. Physiologisch ist FMN von besonderer Bedeutung, da es sowohl an Ein- als auch Zwei-Elektronentransfers partizipieren kann. Es ist ein stärkeres Oxidationsmittel als Nicotinamid-Adenin-Dinukleotid (NAD), ein Coenzym ähnlicher Funktion. Im Rahmen katalytischer Zyklen durchläuft FMN die Formen Semichinon (FMNH•) und die vollends reduzierte Form: FMNH2. FMN ist die vorwiegende Form, in der Flavine in Zellen und Gewebe vorliegt - zwar benötigt der Organismus die Energie
2Umgangssprachlich wird es auch Wachstumsvitamin genannt.
3Der Chromophor ruft einen gelben Farbeindruck hervor (lat.: flavus, zu deutsch: gelb).
2
KAPITEL 1. EINLEITUNG
eines Adenintriphosphats4 (ATP) zur katalytischen Synthese, physiologisch liegt der Vorteil je- doch in der vergleichsweise ausgeprägteren Löslichkeit. Insbesondere die Rolle als Chromophor in Phototropinen geriet in den letzten Jahren in den Blickpunkt, da eine Reihe von photobio- logischen Prozessen auf diese zurückgeführt werden können. Aktuell sind drei unterschiedliche Klassen von Flavin-basierten Photorezeptoren bekannt:
• Phototropine (phot)
• Cryptochrome5 (cry)
• BLUF (sensors of Blue Light Using FAD)
Die beiden letztgenannten Familien basieren auf FAD, Phototropine hingegen verwenden das Mononukleotid als Chromophor. Den Rezeptoren können diverse photobiologische Mechanismen zugeordnet werden; Phototropine regulieren unter anderem folgende Prozesse:
• Phototropie (Pflanzenwachstum gen Lichtquelle, namensgebend).
• Chloroplast-Bewegung (Photorelokation: Phototropine wirken in diversen Pflanzen als Photorezeptoren für die Relokation von Mitochondrien in vergleichsweise schwach be- leuchtete Regionen, um die Photosynthese effizienter zu gestalten; infolge von Signal- transduktion wird die Bewegung von Mitochondrien veranlasst [SW07].
• Stomata-Öffnung: Bei einer Stoma handelt es sich um eine Spaltöffnung (Pore) in der Epidermis von Pflanzen. Sie dient der Regulation des Gasaustauschs der Pflanze mit der Umgebungsluft. Im Allgemeinen bedeutet dies die Aufnahme von Kohlenstoffdioxid (CO2), Abgabe von Sauerstoff (O2) und Transpiration. Die Öffnungsweite der Stoma unter anderem durch die Lichtstärke gesteuert: Mechanistisch handelt es sich um eine Phototropin-vermittelte Signaltransduktion ausgehend von Zellen, die auf den Austausch via Stoma ausgerichtet sind („Guard cells“)[ITS10].
• Inhibition von Stammwachstum [PPS01].
• Gametogenese (Keimzellentwicklung) [HB03].
• etc.
Die erwähnten Prozesse können uniform auf Blaulicht-vermittelte Signaltransduktion zurück- geführt werden. In Phototropinen fungiert dabei FMN als Cofaktor. Im Falle des populärsten Beispiels, dem gerichteten Pflanzenwachstum zur Lichtquelle (Phototropie) kommt es zu fol- gendem photophysikalischen Zyklus: Ausgangspunkt ist ein Addukt aus einem Chromophor -
4Die Domäne der Archaeen („Urbakterien“), die zusammen mit den Bakterien die Einteilung der Prokaryoten bildet, verwendet Cytintriphosphat (CTP) als Energielieferant [SKLP99].
5Der Begriff wurde eingeführt, um die kryptische Natur des Photorezeptors und die kryptogamen Organismen, die in einer Vielzahl in diesem Zusammenhang studiert wurden, als Kofferwort zu vereinen [Gre79].
KAPITEL 1. EINLEITUNG
FMN - und zwei (nichtkovalent gebundenen) Licht-, Sauerstoff- und Spannungs-sensitiven Do- mänen [LOV (light, oxygen, voltage)]. Zunächst unterläuft der Komplex einer Photoanregung, gefolgt von einem schnellen Übergang der Population in den ersten Triplettzustand durch In- terkombination (ISC). In der Konsequenz kommt es im Zuge einer weiteren Interkombination (T S) zur Ausbildung eines metastabilen kovalenten Addukts bestehend aus dem Flavin- komplex und einem Cystein, woraus eine strukturelle Änderung hervorgeht. Diese Signal führt zur Autophosphorylierung. Der Zyklus schließt sich durch Photobleichung - die kovalente Bin- dung zum Cystein spaltet auf und der Chromophor geht in den Grundzustand über [SSJTM09].
Basierend auf diesen Blaulichtrezeptoren wurden spezielle Proteine entwickelt, deren Photo- anregung nicht in Photobleichung resultiert. In der Praxis wurde dazu der Cysteinrest des Apoproteins gegen Alanin ausgetauscht[DEC+07], sodass die Population im ersten angeregten Singulettzustand nicht im Zuge der Konformationsänderung nicht-strahlend verlustig geht und das System zwangsläufig fluoreszent relaxiert. Das Ausschalten des naturgegebenen Photozy- klus durch die Modifikation erhöht die Fluoreszenzintensität erheblich; weitere Verbesserungen im Hinblick auf die Helligkeit [WJDG14] und Photostabilität [CPB+98] konnten durch Muta- genesemethoden erzielt werden6. FMN-bindende Fluoreszenzproteine (FbFP) heben sich durch ihre geringe Größe und Sauerstoffunabhängigkeit von GFP-Derivaten und -Homologen ab - nachteilig gegenüber GFP-basierten Fluoreszenzproteinen ist die deutlich geringer ausgeprägte Helligkeit.
Wie seine Vorläufersubstanz Riboflavin weist FMN eine gelbliche Färbung auf, als solcher findet es industriell ebenso Verwendung als Lebensmittelfarbstoff (E101a). Eine Fortführung des Vit- amin B2-Metabolismus der meisten Tiere ist nach der Bildung von FMN die katalytische Syn- these von Flavin-Adenin-Dinukleotid mittels FMN-Adenyltransferase (FAD-Synthetase) unter Aufwendung eines ATP.
Abbildung 3.: Abbildung des Strukturmotivs von Flavin-Adenin-Dinukleotid (FAD).
6Ebenso konnten die spektralen Eigenschaften verändert werden [WJDG14].
4
KAPITEL 1. EINLEITUNG
FAD dient, ähnlich wie seine Vorläufersubstanz, als Elektronenüberträger in diversen Stoffwech- selprozessen [zum Beispiel: oxidative Phosphorylierung (Acyl-CoA-Dehydrogenase),β-Oxidation von Fettsäuren (Succinat-Dehydrogenase)], dabei ist FAD im Gegensatz zu NAD zum Transfer einzelner Elektronen befähigt. Neben seiner Rolle als Chromophor in diversen Photorezeptoren (cry, BLUF), wirkt FAD vor allem im enzymatischen Kontext [Pyruvatdehy-drogenase-Komplex (PDC; Multienzymkomplex zur oxidativen Decarboxylierung von Pyruvat, stellt das Bindegleid von Glykolyse und Citratzyklus dar), Photolyase (Reparatur UV-geschädigter DNA[TBE+11]), Monoaminoxidase (MAO; mitochondriales Enzym, das dem Abbau giftiger Substanzen dient), etc.]. In folgenden Zusammenhängen spielt FAD als Blaulicht-Photosensor eine elementare Rol- le:
• Taktung der tierischen und pflanzlichen circadianen Rhythmik [vdHH04] (cry).
• Tierische Wahrnehmung des Erdmagnetfeldes [MSWS04] (Magnetsinn) (cry).
• BLUF-Sensoren finden sich in diversen Mikroorganismen im Hinblick auf Signaltransduk- tion wieder [GK02].
Es zeigt sich, dass die Natur Flavin-basierte (Blaulicht-)Photorezeptoren für eine Vielzahl an Funktionen verwendet. Bemerkenswert ist, dass im Kontrast zu allen weiteren bekannten Pho- torezeptoren (Rhodopsine, Xanthopine und Xytochrome) die Signalweiterleitung nicht durch eine Konfigurationsänderung (E/Z-Isomerisation) hervorgerufen wird.
Vorangehend an diese theoretische Studie der fluorierten Flavine wurden diverse experimentelle (zum Beispiel: [SKW+04], [Hee82], [RHTS87]) & theoretische Studien (zum Beispiel: [STM08], [SKW+04], [HBH07], [CGLMSA06], [NSPG03]) zum photophysikalischen und -chemischen Ver- halten von Isoalloxazinen durchgeführt. Die Modifikation der Photophysik infolge von Fluorie- rung am annelierten Benzolring stellt eine vielversprechende Herangehensweise dar, um potente Systeme für die Fluoreszenzmikroskopie und weitere Anwendungen [siehe Kapitel (1.2)] zu ent- wickeln.
KAPITEL 1. EINLEITUNG
1.2. Motivation
Angeschlossen an die Erkundung der Eigenschaften der natürlich vorkommeden Substanz steht die Möglichkeit der chemischen Modifikation des Strukturmotivs. Chemische Substanzen, die gesellschaftliche Anwendung gefunden haben, sind in der Regel der Natur entnommen oder zu- mindest entlehnt. Wissenschaftlich von besonderem Interesse sind sowohl die Eigenschaften der natürlich gegebenen Systeme als auch die Modifikation selbiger, um bestimmte Eigenschaften hervorzurufen und zu modellieren.
Abbildung 4.: Abbildung der Strukturmotive von 6-9F-Methylisoalloxazin.
Der Fokus dieser Arbeit liegt auf der Untersuchung chemisch modifizierter Flavine, als Be- zugspunkt soll dabei stets das Stammmolekül - Methylisoalloxazin (MIA) - dienen. Die Mo- lekülklasse der Flavine nimmt in der Natur eine herausragende Rolle ein, das Strukturmotiv ist evolutionär optimiert, um als Photorezeptor in der Signaltransduktion und bei Redoxpro- zessen in prokaryotischen und eukaryotischen Zellen als Elektronenüberträger zu fungieren.
Die elementare Rolle im Stoffwechsel und der Signaltransduktion [siehe Kapitel 1)] geht einher mit interessanten elektronischen Eigenschaften für potentielle Anwendungen. Durch die Fluo- rierung des annelierten Benzolrings (Positionen 6, 7, 8 und 9; siehe Abbildung 4) können die energetischen Lagen der elektronischen Zustände, die das Molekül einzunehmen vermag und die Eigenschaften definieren, empfindlich verändert werden. Von besonderem Interesse sind da- bei nicht nur die Energien einzelner Zustände, sondern vielmehr, wie diese relativ zueinander stehen. Am Ende der quantenchemischen Untersuchung soll unter anderem festgestellt werden, welche Derivate am besten folgende Funktionen erfüllen können:
• Isoalloxazin eignet sich strukturell in besonderem Maße als Fluoreszenzmarker für photo- biologische Studien/Biodiagnostik ([JG15]; zum Beispiel: FMN-bindende Fluoreszenzpro- teine (FbFP), [DEC+07], siehe auch Kapitel 1), da sich das Stickstoffatom in Position 10 zur definierten Markierung anbietet. Für diese Anwendung ist eine größtmögliche Fluo- reszenzquantenausbeute ΦF anzustreben, um ein möglichst großes Signal bei niedrigen Anregungsintensitäten zu erzielen. Letztere sind von Nöten, da die markierten Systeme
6
KAPITEL 1. EINLEITUNG
selbst nicht aktiviert werden sollen und der Chromophor vor Photobleichung geschützt werden muss.
• Für die Anwendung als Fluoreszenzmarker ist eine möglichst geringe Triplettquanten- ausbeute ΦT wünschenswert, um Nebenreaktionen zu vermeiden und ein großes Signal zu erhalten. Eine potentielle Anwendung hoher Triplettquantenausbeuten stellt die Ge- nerierung von (reaktivem) Singulettsauerstoff dar [SLRD+11], welcher zur Bekämpfung entarteter Zellen (Tumore) genutzt werden kann [Photodynamische Therapie (PDT) [ABC+11]].7
Weitere potentielle Anwendungen sind:
• Photo-Redox Katalyse ([FTT89], [SSK08], [RN16])
• (De-)Sensibilisation in der Fluoreszenzmikroskopie
Die Quantenchemie bietet mittlerweile die Mittel nicht nur einzelne Moleküle zu untersuchen, sondern gleichermaßen die Umgebung dieser einzubeziehen. Die Kombination quantenmecha- nischer Ansätze mit klassicher Physik (Quantenmechanik/Molekülmechanik, QM/MM) ermög- licht die Erforschung von Systemen, die allein quantenchemisch aus Kosten- beziehungsweise Zeitgründen keine Umsetzung findet und daher zuvor einzig experimentell zugänglich war (bei- spielhaft: [SSJTM09], [DW15], [PAW+10]). Zentraler Bestandteil dieser Arbeit ist neben der Fluorierung die Beschreibung der Derivate in wässrigem Medium - sowohl die intramolekulare Modifikation als auch externe Beeinflussung gilt es zu untersuchen, um das real zu erwartende Resultat beschreiben zu können.
7Neben einer hohen ΦT ist eine vernachlässigbare Toxizität des Photosensibilisators von Nöten, sodass die Substanz einem Patienten (lokal oder systemisch) verabreicht werden kann.
Teil I.
Theorie & Praxis
2. Theoretischer Hintergrund
In diesem Kapitel aufgeführtes Wissen ist zum Großteil Gegenstand der Hochschullehre.
2.1. Motivation der Schrödingergleichung
Die Postulierung der Schrödingergleichung stellt die Geburtsstunde der Quantenchemie dar.
Einst wurde sie herangezogen, um das Linienspektrum des Wasserstoffatoms zu erklären, im Laufe der Zeit hat sich ein versierter Wissenschaftszweig empor gebildet. Formal handelt es sich bei der fundamentalen Gleichung um ein Postulat - sie kann nicht aus der klassischen Mechanik hergeleitet werden. Die Schrödingergleichung erfasst die Energie eines beliebigen Systems auf quantenmechanischer Ebene, das heißt ihre Beschreibung findet auf Basis des Welle-Teilchen- Dualismus statt. Charakteristika von Materie sind zum einen eine definierte Masse, zum anderen gehorcht sie dem Prinzip der (Dreh-)Impulserhaltung. Der experimentelle Befund schreibt die- se Eigenschaften zwar auch mikroskopischen Teilchen wie Photonen und Elektronen zu, jedoch haben diese gleichermaßen Welleneigenschaften, beispielsweise lässt sich das Phänomen der Beugung feststellen. Die klassische Mechanik vermag diese Eigenschaften, den Dualismus, nicht vereinheitlicht zu beschreiben. Quantenmechanisch erfolgt die Beschreibung des Teilchens als Wellenfunktion Ψ mit Impuls sowie Masse und an Stelle klassischer Größen stehen korrespon- dierende Operatoren (Korrespondenzprinzip). Unmittelbar ergibt sich die Schrödingergleichung somit durch Austausch der Größen in der Hamiltonfunktion H:
H=Etot =Ekin+Epot = ~p2
2m +V(~r, t) (1) Das Korrespondenzprinzip besagt, dass die Gesamtenergie E, der Impuls ~p sowie Potential V (Ort~r) durch die quantenmechanischen Gegenstücke substituiert werden können:
• E→Eˆ =i~∂t∂
• ~p→pˆ=−i~∇
• ~r→~rˆ=~r
• V(~r, t)→Vˆ(~r, t)
KAPITEL 2. THEORETISCHER HINTERGRUND
Somit ergibt sich unmittelbar die zeitabhängige Schrödingergleichung:
i~
∂Ψ
∂t =−~2
2m∆Ψ + ˆV(~r, t)Ψ = ˆH (2)
In der Folge wird eine anschauliche Herleitung dargelegt, die explizit die Zusammenführung von Teilchen- und Wellencharakter - den Dualismus - beschreibt. Gemäß De Broglie kann jedem Teilchen eine Wellenlänge zugeordnet werden:
~
p=~~k (3)
Aufkommen von Impuls ist somit unmittelbar mit dem Dasein einer Welle verknüpft, einer so- genannten Materiewelle. Der Impuls ist durch den Wellenvektor~k definiert, welcher die gleich- förmige Ausbreitungsrichtung angibt und den Betrag k = 2πλ aufweist. Unter der Annahme, dass die Materiewelle der Wellengleichung gehorcht, kann eine Beschreibung dieser innerhalb der Raumzeit gefunden werden:
∆Ψ =c−2∂2Ψ
∂t2 (4)
Mit c = λ · ν, |k| = 2πλ sowie ω = 2πT = 2π ·ν lassen sich folgende reelle Lösungen der Differentialgleichung heranziehen:
Ψ1(~r, t) = Ψ0cos(ωt−~k~r+φ)∧Ψ2(~r, t) = Ψ0cos(ωt+~k~r+φ) (5) Beide Lösungen lassen sich wiederum zu einem Produktansatz vereinigen.1
Ψ(~r, t) = Ψ0
2 [cos(ωt−~k~r) +cos(ωt+~k~r)] = Ψ0sin(ωt)·sin(~k~r) =φ(~k~r)·sin(ωt) (6) Durch Einsetzen dieser Lösung in die Wellengleichung (Gleichung 4) ergibt sich:
∆φ+ω2
c2φ = ∆φ+ (2πν
c )2φ= 0 (7)
Umformung des konstanten Parts mit c = ν ·λ und der De-Broglie-Beziehung p = hλ ergibt folgenden Faktor:
2πν c
2
=
2π λ
2
= 2π~p h
!2
= ~p
~
!2
(8) Ausdruck (7) geht somit über in:
∆φ+ (p~
~
)2φ = 0 (9)
1Dieser ist an die Bedingung geknüpft, dass das betrachtete System keinem zeitabhängigen Potential ausgesetzt wird.
12
KAPITEL 2. THEORETISCHER HINTERGRUND
Der klassische dreidimensionale Ausdruck für die kinetische Energie lautet:
Ekin= m~v2 2 = ~p2
2m (10)
Einsetzen in Gleichung 9 liefert den kinetischen Energieoperator:
− ~2
2m∆φ−Ekinφ= 0⇐⇒ − ~2
2m∆φ=Ekinφ (11)
Mit der Hamiltonfunktion erhält man folgerichtig die zeitunabhängige Schrödingergleichung:
−~2
2m∆φ−(Etot−Epot) = 0⇐⇒ − ~2
2m∆φ+Epot=Etot (12) Diese beschreibt stationäre Zustände. Die Gleichung bezieht sich somit auf eine unendliche Lebensdauer. Zwar ist dies utopisch, da vor allem angeregte Zustände auf menschlich sehr kur- zen Zeitskalen relaxieren, jedoch können mit Hilfe der zeitunabhängigen Schrödingergleichung
„Übergangsstationen des elektronischen Energiekreislaufs“ konkretisiert werden. Für die Herlei- tung der zu Beginn dargelegten zeitabhängigen Version wird noch ein weiterer Operator für die Gesamtenergie benötigt [siehe Gleichung (2)]. Betrachten wir nun eine komplexwertige Lösung der Wellengleichung:
Ψ = Ψ0ei(~k~r−ωt) (13)
Diese lässt sich mit E =h·ν und c=ν·λ umschreiben:
Ψ = Ψ0ei/~(~p~r−Et) (14)
Die zeitliche Ableitung liefert den Energieeigenwert der Eigenfunktion zui~∂Ψ∂t, welcher schluss- richtig den gesuchten Energieoperator darstellt.2
∂Ψ
∂t =−iE
~ Ψ0ei/~(~p~r−Et) ⇐⇒EΨ =i~∂Ψ
∂t (15)
Das (vernachlässigbare) zeitliche Verhalten der zuvor beschriebenen stationären Eigenfunktio- nen ist somit trigonometrisch beschreibbar - als periodische Schwankung. Wird das System jedoch einem zeitabhängigen Potential ausgesetzt, so entwickelt sich die Gesamtenergie zeitlich analog zur Störung.
Im Hinblick auf die Hamiltonfunktion können resümierend die benötigten, mit dem Dualismus Teilchen-Welle in Einklang stehenden Operatoren, aus der Quantenmechanik via Wellenglei- chung erhalten und korrespondierend für die klassichen Größen eingesetzt werden.3 Bezogen
2Analoges Vorgehen in der räumlichen Domäne liefert den Impulsoperator, welcher wiederum gemäß Gleichung (10) direkt in den kinetischen Energieoperator überführt werden kann.
3Die Willkür der Lösung der Wellengleichung/einer Differentialgleichung ermöglicht dabei, wie beschrieben, unterschiedliche Ansätze (beispielsweise reell/komplexwertig).
KAPITEL 2. THEORETISCHER HINTERGRUND
auf die ursprüngliche Problemstellung der Deutung des Linienspektrum von Wasserstoff, be- schreibt jede Eigenfunktion des Hamiltonoperators ein Orbital mit dem dazugehörigen Ener- gieeigenwert. Nach Max Born ist das Betragsquadrat der normierten Wellenfunktion äquivalent zur Aufenthaltswahrscheinlichkeit des beschriebenen Teilchens (hier: Elektron im Potential des Protons) im bezüglichen Raumelement d~r.
2.2. Das Abklingen angeregter Zustände
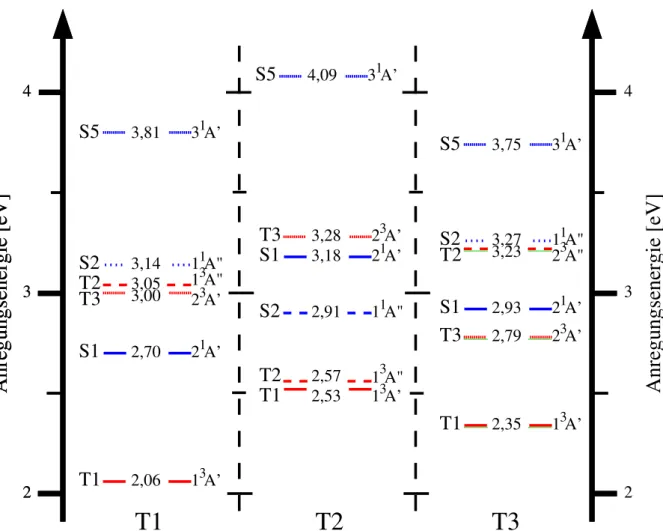
Abbildung 5.: Jabłoński-Termschema; vibronische Zustände und mögliche Angerungs- sowie Relaxationspfade eines organischen Chromophors. Die gestrichelten, horizonta- len Pfeile illustrieren Interkombination (ISC) beziehungsweise interne Konversion (IC). Prozesse, die unter Absorption respektive Emission eines Photons ablaufen, sind durch geradlinige, vertikale Pfeile angedeutet (A:Absorption, F: Fluores- zenz, P: Phosphoreszenz). Die wellenförmigen Pfeile stellen Stoßrelaxation (VR) dar [Bra15].
Trifft Strahlung auf ein Molekel, so wird dieses gemäß Energieerhaltung in einen angeregten Zu- stand gebracht. Ein Zustand ist allgemein rovibronischer Natur, das bedeutet er ist aus einem elektronischen, Rotations- und Schwingungszustand komponiert. Die Energiedifferenzen von Rotationszuständen sind für die im Rahmen dieser Arbeit betrachteten Prozesse vernachlässig- bar; in der Folge werden photoinduzierte Prozesse vibronischer Zustände erläutert, prinzipiell lassen sich für Schwingungszustände angestellte Gedanken jedoch direkt auf Rotationszustände übertragen.
14
KAPITEL 2. THEORETISCHER HINTERGRUND
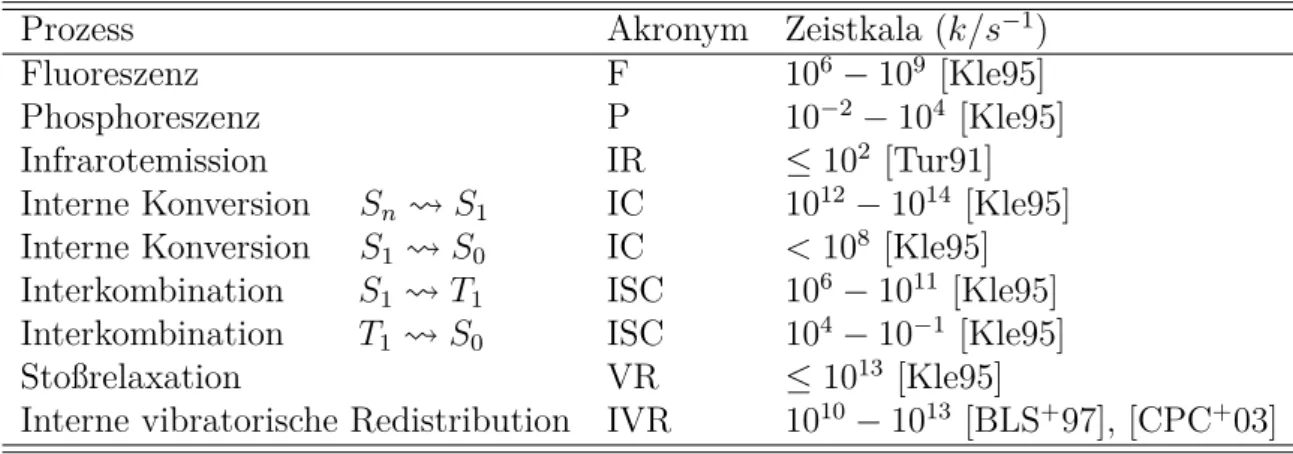
Im Falle einer resonanten vibronischen Anregung eines Moleküls dissipiert in der Folge die absorbierte Energie. Dies kann über diverse Kanäle geschehen, die sich allgemein in strahlende und strahlungslose Übergänge einteilen lassen. Folgende Tabelle stellt die wichtigsten photo- physikalische Folgeprozesse der Absorption dar und ordnet diesen eine Zeitskala zu:4
Tabelle 1.: Zusammenstellung der Zeitskalen für diverse photophysikalische Folgeprozesse der Absorption mitsamt konventioneller Akronyme.
Prozess Akronym Zeistkala (k/s−1)
Fluoreszenz F 106−109 [Kle95]
Phosphoreszenz P 10−2−104 [Kle95]
Infrarotemission IR ≤102 [Tur91]
Interne Konversion Sn S1 IC 1012−1014 [Kle95]
Interne Konversion S1 S0 IC <108 [Kle95]
Interkombination S1 T1 ISC 106−1011 [Kle95]
Interkombination T1 S0 ISC 104−10−1 [Kle95]
Stoßrelaxation VR ≤1013 [Kle95]
Interne vibratorische Redistribution IVR 1010−1013 [BLS+97], [CPC+03]
Im Falle strahlender Übergänge (Lumineszenz) folgt auf die Absorption eines Photons die Emis- sion. Fluoreszenz (F) bezeichnet dabei in der Regel die Strahlungsabgabe vom schwingungs- losen S1-Zustand in Schwingungszustände des Grundzustands S0. Phosphoreszenz (P) findet im Gegensatz zur Fluoreszenz unter Spinumkehr statt und ist somit ein verbotener Übergang.
Nicht-strahlende elektronische Übergänge können gleichermaßen unter Spinumkehr oder -erhalt stattfinden. Der verbotene Prozess wird als Interkombination [Intersystem Crossing (ISC)] be- zeichnet. Das ohne Spinumkehr auskommende Gegenstück ist die Interne Konversion [Internal Conversion (IC)]. Im Anschluss an ISC und IC befindet sich das Molekül aufgrund der un- mittelbaren Energieerhaltung in hochangeregten Schwingungszuständen. Die Reduktion der Schwingungsenergie einer Mode erfolgt zum einen durch die Interaktion mit der Umgebung [Vibrational Relaxation (VR)] und - dem Energieübertrag auf deren Freiheitsgrade. Andern- falls kann sich auch intramolekular eine Fluktuation zwischen Moden [Internal Vibrational Energy Redistribution (IVR)] ereignen. Neben der Lumineszenz, die sich im Allgemeinen auf den menschlich sichtbaren Spektralbereich (VIS: circa 380−780nm) bezieht, kann gleicher- maßen Schwingungsenergie als Wellenpaket des Infrarotbereichs [Infrarotstrahlung (IR: circa 780nm−1mm)] emittiert werden.
Jeder Folgeprozess hat eine intrinsische Zeitskala, die unmittelbar mit der Lebensdauer der betroffenen Zustände verknüpft ist. Die Lebensdauer eines beliebigen Ausgangszustands eines
4Ausführungen werden in der Folge auf Singulett- und Triplettzustände beschränkt. Höhere Multiplizitäten können im Rahmen dieser Arbeit vernachlässigt werden, verhalten sich prinzipiell jedoch ähnlich
KAPITEL 2. THEORETISCHER HINTERGRUND
Folgeprozesses der Anregung ist wie folgt definiert und mit der Ratenkonstante k verknüpft:5 τX = 1
kX = 1
n
X
i
ki
(16)
Demzufolge ist die Lebensdauer τX eines Zustands X durch die Summe der Prozesse n, die X depopulieren, bestimmt. Die reziproke Lebensdauer entspricht somit der Ratenkonstante mit welcher der Zustand abklingt (kX).
Fluoreszenzlebensdauern zum Beispiel korrespondieren in aller Regel mit der Lebensdauer des S1-Zustands und belaufen sich auf Piko- bis Nanosekunden. Das verbotene Pendant, Phospho- reszenz (T1) findet im Mikrosekundenbereich bis hin zu einigen Minuten statt. Die Geschwin- digkeit und somit Lebensdauer der Internen Konversion und Interkombination ist abhängig vom betroffenen Zustand: DieEnergy Gap Rulebesagt, dass die Ratenkonstante umgekehrt propor- tional zur Energielücke der betrachteten Zustände ist (exponentielle Abnahme). Dementspre- chend benötigt die Interne Konversion von Energie aus einem n-ten Singulettzustand in den ersten lediglich Femto- bis Pikosekunden, wohingegen der S1 S0-Übergang in weniger als einer Nanosekunde vollzogen ist. Interkombination findet aufgrund des involvierten Übergang- verbots in der Regel6 auf einer deutlich längeren Zeitskala statt; aus demS1- in denT1-Zustand kann sie Mikrosekunden dauern, ISC aus letzterem in den Grundzustand sogar Sekunden.7 An dieser Stelle sei anzumerken, dass Interkombination ebenso über intermediäre Zustände statt- finden kann, beispielsweise wie folgt:S1 Tn T1. IVR und VR finden auf derselben Zeitskala wie Schwingungen statt (. 100f s), da es in beiden Varianten zum Energieübertrag zwischen den jeweilig beteiligten Moden kommt.
Ob ein einzelner Prozess i detektiert werden kann, hängt mit seiner Quantenausbeute Φi (sie- he Gleichung 17) (und Empfindlichkeit der Messtechnik) zusammen. Jeder Prozess, der zwei Zustände ineinander überführt, konkurriert mit weiteren, sodass vergleichsweise langsame Pro- zesse auch relativ selten geschehen, ehe der Ausgangszustand depopuliert ist.
Φi = ki
n
X
i
ki
(17)
Die Quantenausbeute eines Prozesses Φi ist ein Maß für den Anteil eines Prozesses i am Ab- klingen eines Zustands. Zwar haben Fluoreszenz und Phosphoreszenz auch einen Anteil an der Depopulation höherer Zustände als S1/T1, jedoch ist dieser normalerweise zu gering, um de- tektiert zu werden. Ausnahmen gegen die Kasha-Regel [Kas50], die besagt, dass die Emission eines Photons aus dem niedrigsten angeregten Zustand einer gegebenen Multiplizität stammt, sind beispielsweise Azulen (F aus S2) [Tur91] und Dimethylbenzaldehyd (P aus T2) [DMB76].
5An dieser Stelle wird sich auf Energietransfer mit Ratenkonstanten erster Ordnung beschränkt.
6Ausnahmen stellen beispielsweise aromatische Carbonyle (etwa Thioxanthon) dar [ZFG+17].
7Im Zuge thermischer Aktivierung ist zudem Interkombination aus demT1- in denS1-Zustand möglich (TADF, [LM17]). Dieser Vorgang wird auch als Re-ISC bezeichnet.
16
KAPITEL 2. THEORETISCHER HINTERGRUND
2.2.1. Helle und dunkle Zustände
Im Experiment können lediglich bestimmte Zustände adressiert werden. Die Möglichkeit ei- nes Zustands, durch Absorption populiert zu werden, ist abhängig vom Übergangsmoment:
Experimentell muss ein Singulettzustand mit einem ausgeprägten Moment ausfindig gemacht werden, in der Folge kann der Experimentator jene Zustände, die im Zuge der Relaxation des Systems durchlaufen werden, detektieren und sich ein Bild von der elektronischen Struktur machen. Dabei kann sich darüber hinaus zu Nutze gemacht werden, dass die spektroskopischen Auswahlregeln je nach Experiment variieren (zum Beispiel: Resonanz-Raman-, 2-Photonen-, Anionen Photodetachment Photoelektronen-Spektroskopie).
Die Terminologie hell und dunkel bezieht sich auf das elektrische Übergangsdipolmoment re- spektive die Oszillatorstärke eines Übergangs. Zur Beleuchtung dunkler Zustände ist es häufig praktisch auf quantenchemische Rechnungen zurückzugreifen - auf diese Weise können Spektren detailliert verstanden werden. Das elektrische Übergangsdipolmoment ist definiert als Integral des Anfangs- und Endzustandsi f über den elektrischen Übergangsdipoloperator ˆ~µ(Neben- diagonalelement des Dipolmomentoperators):
~
µf i =hΨf|~µ|ϕˆ ii (18) Das Betragsquadrat des Übergangsdipolmoments ist proportional zur Übergangswahrschein- lichkeit/Oszillatorstärke. Letztere Größe stellt eine Verbindung zwischen experimentell beob- achteter Bande und Berechnung her [Hak06]:
f = 8π2me−
3he2 |hϕf|~µ|Ψˆ ii|2·ν (19)
= 40me−ln(10)c e2NA ·
Z
(˜ν)d˜ν (20)
KAPITEL 2. THEORETISCHER HINTERGRUND
2.3. Dichtefunktionaltheorie (DFT)
Die im Bereich der theoretischen Chemie eingesetzte Dichtefunktionaltheorie hat formal be- trachtet (Kohn-Sham-Formalismus) große Ähnlichkeiten mit dem Hartree-Fock-Verfahren. Die prinzipielle Idee hinter diesem ist die Lösung der zeitunabhängigen Schrödinger-Gleichung im Kontext der Born-Oppenheimer-Näherung. Dazu wird die Wellenfunktion als Slaterdeterminan- te8 angesetzt und die Minimierung der Energie im Rahmen der SCF-Näherung [self-consistent (mean) field] mittels Variationsprinzip durchgeführt. Das einwirkende Potential ist durch den Hamiltonoperator ˆH gegeben.9
Hˆ =− ~2 2me
n
X
i
∇2i −
n
X
i N
X
I
ZIe2 4π0rIi +1
2
n
X
ij
e2
4π0rij (21)
- es resultieren gekoppelte Integro-Diffenrentialgleichungen, die iterativ gelöst werden müssen.
Dies geschieht durch Verwendung eines festgelegten Basissatz - optimiert werden die Koeffizi- enten zur Gewichtung der jeweiligen Basisfunktionen bis zur Selbstkonsistenz (SCF).
Im Gegensatz zu Wellenfunktions-basierten ΨN Methoden ist in der Dichtefunktionaltheorie (DF T) die Elektronendichte ρ(~r) die zentrale Größe. Grundgedanke hinter der Substitution ist die Reduktion der Koordinaten - trotz eines N-Elektronensystems sind lediglich drei Koordi- naten von Nöten, um die umfassende Elektronendichte zu beschreiben. Formell wird diese als Wegintegral eines Elektrons des N-Elektronensystems über die Koordinaten der mitstreitenden Elektronen beschrieben. Per Definition muss das Integral der Dichte über den gesamten Raum gleich der Elektronenanzahl n sein:
ρ(~r) =n
Z
...
Z
|Ψ(d~x1d~x2...d~xn)|2ds1d~x2...d~xn (22) Die Hohenberg-Kohn-Theoreme (HK-Theoreme) liefern den Leitfaden, um mittels dieser Defini- tion ein Elektronendichte-Analogon zum Hartree-Fock-Verfahren herzuleiten. Sie beziehen sich dabei auf einen nicht-entarteten Grundzustand. Das erste Theorem besagt, dass zwischen dem externen Potential υ(~r) und der Elektronendichte ρ(~r) eine direkte Korrespondenz besteht. In der Konsequenz kann aus der Dichte das externe Potential (bis auf eine additive Konstante) ab- geleitet werden. Umformuliert, in Anbetracht des Gesichtspunkts ausgeschlossener Entartung, verlautbart der erste Leitsatz, dass die von den Hamiltonoperatoren ˆH und ˆH0 abgeleiteten Wellenfunktionen Ψ, Ψ0 nicht zur selben Elektronendichteρ=ρ0 gehören können.
1. Hohenberg-Kohn Theorem
Die Elektronendichte ρ(~r) bestimmt das externe Potential.
8Deren Funktion besteht in der Antisymmetrisierung der Wellenfunktion, sodass diese Fermionen (Elektronen) repräsentieren kann.
9Die Repulsion der N Kerne untereinander wird zusätzlich addiert.
18
KAPITEL 2. THEORETISCHER HINTERGRUND
Dementsprechend darf die Energie als Funktional der Dichtefunktion ρ(~r) wie folgt zusam- mengesetzt werden:
E[ρ] =
Z
ρ(~r)υ(~r)d~r+
n
X
i
hφi| − 1
2∇2|φii+Vel,el[ρ] (23)
=Vnuc,el[ρ] +T[ρ] +Vel,el[ρ] (24) In der dargelegten Summation beschreibt Vnuc,el die Kern-Elektron-Wechselwirkung - das ex- terne Potential ist somit das Kernpotentialυ.T steht für die kinetische Energie der Elektronen und Vel,el umfasst die repulsive Wechselwirkung der Elektronen untereinander, bestehend aus der klassischen Coulomb-Wechselwirkung J[ρ] [siehe Formel (31)] und der nicht-klassischen Austauschwechselwirkung (K).
Das zweite Hohenberg-Kohn Theorem besagt, dass analog zu wellenfunktionsbasierten Ver- fahren ein Variationsprinzip auf die Elektronendichte zur Minimierung der Gesamtenergie des Systems E[ρ] angewandt werden kann.
2. Hohenberg-Kohn Theorem
Die Grundzustandsenergie Eexakt kann variationell erhalten werden: Die Dichte ˜ρ, welche die totale EnergieE minimiert, ist die exakte Grundzustandsdichte ρ.
Somit gilt für einen beliebigen Testansatz der Elektronendichte ˜ρ:
E[ ˜ρ] =
Z
Vnuc,el[ ˜ρ] +T[ ˜ρ] +Vel,el[ ˜ρ]≥Eexakt (25) Der zweite Leitsatz eröffnet die Möglichkeit, Verfahren zur Minimierung der Gesamtenergie eines Systems zu konzipieren. Im quantenchemischen Kontext ist an dieser Stelle vor allem der auf dem Orbitalkonzept basierende Kohn-Sham-Formalismus zu nennen. Das Konzept sieht vor, dass zunächst ein fiktives System bestehend aus N Fermionen ohne wechselseitige Interaktion definiert wird. Die Pseudo-Elektronen weisen weder Ladung noch Spin auf, gehorchen aber den- noch dem Pauli-Prinzip. Das System wird mithilfe dieser Hilfsvariable durch eine Wellenfunk- tion in Determinanten-Form zusammengesetzt aus N Elektronen in N Kohn-Sham-Orbitalen φi repräsentiert. Die Elektronendichte für ein derartiges System lautet wie folgt:
ρ=
N
X
i
|φi(~r)|2 (26) Die einzelnen KS-Orbitale und dazugehörigen Energien gehorchen analog zur Hartree-Fock- Methodik folgenden Euler-Lagrange-Gleichungen:
−1
2∇2+υs(~r)
φi(~r) = iφi(~r) (27)
KAPITEL 2. THEORETISCHER HINTERGRUND
Die Gesamtenergie des fiktiven Systems setzt sich analog zu Gleichung (24) aus zwei Termen zusammen - der kinetischen EnergieTs und der Kern-Elektron-Wechselwirkung Vs (=Vnuc,el):
E[ρ] =Vs[ρ] +Ts[ρ] (28)
Ausgehend von dieser Energie für das fiktive System ergibt sich für das zu beschreibende reale Pendant durch Hinzuziehen eines Terms Vel,el:
E[ρ] =Vs[ρ] +Ts[ρ] +Vel,el[ρ] (29)
=Vnuc,el[ρ] +T[ρ] +J[ρ] +Exc[ρ] (30) Aus Gleichung (30) geht hervor, dass die Coulomb-Abstoßung der ElektronenVel,elin die Terme J (klassisch) und Exc (nicht-klassisch) aufgetrennt wird. Die Austauschenergie (Exc) beinhal- tet zudem die Differenz T −Ts. Die Coulomb-Wechselwirkung kann über die Gesamtheit der Elektronen, die Dichte, beschrieben werden:
J[ρ] = 1 2
Z Z ρ(~r)ρ(~r0)
|~r−~r0| d~rd~r0 (31) Zusammenfassend ergeben sich die Eigenfunktionen und -werte folgendes Kohn-Sham-Opera- tors ˆfKS zur Beschreibung optimaler Orbitale sowie Orbitalenergien:
fˆKSφi(~r) =
"
−1
2∇2+υ(~r) +
Z ρ(~r0)
|~r−~r0|d~r0+υxc(~r)
#
φi(~r) = iφi(~r) (32) Die Orbitale werden in der Konsequenz, analog zum Vorgehen im Rahmen der HF-Theorie, in einem Basissatz expandiert, woraus wiederum KS-SCF-Gleichungen resultieren.
Das Austausch- Korrelationspotential υxc entspricht der Ableitung der Austauschenergie Exc nach der Elektronendichte,
υxc(~r) = δExc
δρ(~r, t) (33)
es beschreibt sowohl den energetischen Anteil der Austauschwechselwirkung (x) als auch die Be- wegung der jeweiligen Elektronen im gemittelten Feld der anderen. Die Kohn-Sham-Gleichungen (32) sind den kanonischen Hartree-Fock-Gleichungen strukturell sehr ähnlich, allerdings unter- scheidet sich das Potentialυxcin grundlegender Weise vom wellenfunktionsbasierten Austausch- operator ˆK- das Austausch-Korrelationspotential muss approximiert werden und ist konzipiert, um neben der Austauschwechselwirkung die Elektronenkorrelation (c) zu umfassen. In der Pra- xis unterscheidet man je nach Beschreibung (Ψ/ρ) der kinetischen EnergieET undExczwischen folgenden Herangehensweisen:
• HF-basierte Theorie: ET ←Ψ und Exc←Ψ
• Adiabatic Connection (AC):ET ←Ψ und Exc←Ψ, ρ
20
KAPITEL 2. THEORETISCHER HINTERGRUND
• KS-basierte Theorie: ET ←Ψ undExc ←ρ
• Alleinige DFT: ET ←ρ und Exc←ρ
Im Rahmen dieser Arbeit kommen die AC-(Hybrid-)Funktionale B3LYP sowie BHLYP zum Einsatz.
Zwar ergibt sich durch die bisher dargelegte Theorie einzig eine Aussage über den Grund- zustand, jedoch lässt sich der Ansatz um die Zeitdomäne erweitern (Time-Dependent-DFT/
TDDFT), sodass gleichermaßen angeregte Zustände beschrieben werden können. Prinzipiell wird hierbei die lineare Antwort (Response) des Systems im Grundzustand auf eine zeitabhän- gige Störung (z. B. externe elektromagnetische Felder) untersucht. Die Anregungsenergie in Form einer Frequenz (ω) ergibt sich hierbei als die Polstelle der frequenzabhängigen Polarisier- barkeit.
2.3.1. Zeitabhängige Dichtefunktionaltheorie (TD-DFT)
Die Korrespondenz der Elektronendichte ρ(~r) und des Potentials wird für den Grundzustand durch das erste Hohenberg-Kohn (HK) Theorem dargelegt. Selbiger Zusammenhang muss für die zeitabhängige Beschreibung gegeben sein, damit vom Kernpotential zuzüglich der externen Störung auf die Dichte und folglich auf die (Anregungs-)Energien sowie Orbitale geschlossen werden kann. Das zeitabhängige Analogon zum ersten HK-Theorem ist das Runge-Gross Theo- rem [RG84]. Dieses besagt, dass die exakte zeitabhängige Elektronendichte ρ(~r, t) das zeitab- hängige externe Potential V(~r, t) bis auf eine räumlich konstante, zeitabhängige FunktionC(t) beschreibt. Daraus ergibt sich unmittelbar die Korrespondenz beider mit der dazugehörigen zeitabhängigen Wellenfunktion Ψ(~r, t) als Funktional von ρ, zuzüglich eines zeitabhängigen Phasenfaktors:
ρ(~r, t)↔υ[ρ](~r, t) +C(t)↔Ψ[ρ](~r, t)e−iα(t) (34) Runge-Gross Theorem
Die zeitabhängige Elektronendichte ρ(~r, t) bestimmt das zeitabhängige externe Potential.
Trotz des Phasenfaktors e−iα(t) ist der Erwartungswert eines beliebigen quantenmechanischen Operators ein Funktional der Dichte, da der Faktor multipliziert mit seiner Adjungierten eli- miniert wird [DHG05]. Das zeitabhängige Variationsprinzip kann nicht äquivalent zum zweiten Hohenberg-Kohn Theorem formuliert werden, da zunächst ein stationärer Punkt ausgemacht werden muss. Dies erfolgt unter Verwendung des quantenmechanischen AktionintegralsA, wel- ches basierend auf dem Runge-Gross Theorem als Funktional A[ρ] formuliert wie folgt lautet:
A[ρ] =
t1
Z
t0
dthΨ[ρ](~r, t)|iδ
δt−H(~ˆ r, t)|Ψ[ρ](~r, t)i (35)
KAPITEL 2. THEORETISCHER HINTERGRUND
Eigenzustände des Systems können ausfindig gemacht werden, indem das Integral unter Varia- tion der Elektronendichte auf folgende Bedingung (Euler-Gleichung) hin überprüft wird:
δA[ρ]
δρ(~r, t) = 0 (36)
Auf diese Weise ist die exakte Dichteverteilung eines Zustands in der Zeitdomäne zugänglich.
Darauf gründend kann analog zu den zeitunabhängigen - per se stationären - KS-Gleichungen ein zeitabhängiges Pendant konstruiert werden. Dementsprechend werden zur Beschreibung des Zustands unter der Bedingung, dass die Elektronen miteinander keine Wechselwirkung einge- hen [(siehe Gleichung (26)], Einelektronen-Orbitale definiert. Die entsprechenden Ein-(Pseudo- )Elektronen-Orbitale resultieren aus folgender zeitabhängigen Schrödingergleichung [vergleiche Gleichung (27)]:
−1
2∇2+υs(~r)
φi(~r) = iδ
δtφi(~r) (37)
Die analogen KS-Gleichungen ergeben sich unter der Annahme, dass die Dichte der Pseudo- Elektronen dieselbe Beschaffenheit wie die exakte hat. Unter Verwendung des Aktionintegrals kann somit folgender Ausdruck hergeleitet werden.
fˆKSφi(~r, t) =
"
−1
2∇2+υ(~r, t) +
Z ρ(~r0, t)
|~r−~r0|d~r0+ δAxc[ρ δρ(~r, t)
#
φi(~r) =iδ
δtφi(~r) (38) Diese unterscheiden sich einzig durch den Austausch-Korrelationsterm (xc) und die zeitliche Ab- leitung multipliziert mit der imaginären Einheit i von den Grundzustandsgleichungen [(siehe Gleichung (32)]. In der Praxis werden jedoch anstatt des Ausdrucks Axc dieselben Funktiona- le wie für den Grundzustand verwendet [DHG05], sodass auf dessen Beschaffenheit an dieser Stelle nicht genauer eingegangen wird.
Für einen beliebigen Basissatz ergibt sich aus Gleichung (38) folgende Schreibweise, wobei C die zeitabhängigen Koeffizienten der Expansion im Basisatz beinhaltet:
FKSC=iδ
δtC (39)
FKS ist hierbei die Matrix-Schreibweise des zeitabhängigen KS-Operators. Via Multiplikation mit C† von rechts und Subtraktion der hermitischen Transponierten lässt sich Gleichung (18) in folgende Dichtematrix/Dirac-Form umschreiben:
X
q
FpqPqr−PpqFqr =iδ
δtPpr (40)
Setzt man das System nun einer oszillierenden, zeitabhängigen Störung aus, so lässt sich die resultierende Dichtematrix für das gestörte System als Summe des ungestörten Bestandteils und der Änderung erster Ordnung (linear Response) formulieren:
Ppq =Ppq(0)+Ppq(1) (41)
22
KAPITEL 2. THEORETISCHER HINTERGRUND
Selbiges gilt für den KS-Hamiltonian:
Fpq =Fpq(0)+Fpq(1) (42) In der Folge werden die Ausdrücke (41) und (42) in Gleichung (40) eingesetzt und die Terme erster Ordnung gesammelt.
X
q
Fpq(0)Pqr(1)−Ppq(1)Fqr(0)+Fpq(1)Pqr(0)−Ppq(0)Fqr(1) =iδ
δtPpr(1) (43) Die Änderung erster Ordndung des KS-Hamiltonians besteht zum einen aus der betrachteten Störung gpq (fpq ist ein Ein-Elektronen-Operator und beschreibt die Störung im Detail):
gpq = 1
2[fpqe−iωt+fqp∗e−iωt] (44) Zum anderen reagiert der Zwei-Elektronen-Part von Fpq(0) auf die Änderung der Dichtematrix.
Diese wiederum wird analog zu Gleichung (44) beschrieben (dpq repräsentiert die Dichten der Störung):
Ppq(1) = 1
2[dpqe−iωt+d∗qpe−iωt] (45) Setzt man nun die Ausdrücke für die jeweiligen Terme Ppq(0/1) und Fpq(0/1) in Gleichung (22) ein, so lässt sich diese auf zwei Ausdrücke reduzieren,10 welche wiederum eine nicht-hermitesche Eigenwert-Gleichung bilden (TDDFT-Gl.):
"
A B
B∗ A∗
# "
X Y
#
=ω
"
1 0
0 −1
# "
X Y
#
(46) Die Matrixelemente A und B sind abhängig vom eingesetzten Funktional; für ein reines Dich- tefunktional sind sie in Mulliken-Notation gegeben durch [DHG05]:
Aia,jb =δijδab(a−i) + (ia|jb) + (ia|fxc|jb) (47) Bia,jb = (ia|jb) + (ia|fxc|jb) (48) Hierbei repräsentierenaundiun-/besetzte Orbitale.AundB unterscheiden sich lediglich durch die Differenz derer Orbitalenergienasowiei, der Zweielektronen-Term und die lineare Antwort des verwendeten Austausch-Korrelation-Funktionals sind identisch. X und Y stellen Amplitu- den dar, welche die An- und Abregung relativ zur Referenzdeterminante beschreiben.ωumfasst die begehrten Eigenwerte, die Anregungsenergien/-frequenzen. Für Hybrid-Dichtefunktionale
10Die Idempotenz-BedingungX
q
Ppq(0)Pqr(0) =Pprstellt eine starke Einschränkung fürdpq[siehe Gleichung (45)]
dar. Desweitern kann die diagonale Beschaffenheit der ungestörten KS-Fock- und Dichtematrizen ausgenutzt werden [DHG05].