Grenzflächenreaktionen von Actiniden an Muskovit
D I S S E R T A T I O N
zur Erlangung des akademischen Grades
Doctor rerum naturalium (Dr. rer. nat.)
vorgelegt
der Fakultät Mathematik und Naturwissenschaften der Technischen Universität Dresden
von
Diplom-Geologe, Stefan Hellebrandt
Geboren am 10.02.1986 in Gera
Eingereicht am 11.08.2017
Die Dissertation wurde in der Zeit von Dezember 2013 bis Juli 2017 im Institut für Ressourcenökologie
Des Helmholtz-Zentrums Dresden-Rossendorf angefertigt.
Gutachter: Prof. Dr. Thorsten Stumpf (TU Dresden) Prof. Dr. Thorsten Schäfer (FSU Jena)
Tag der Verteidigung: 06.11.2017
Danksagung
Die vorliegende Doktorarbeit wurde von Dezember 2013 bis Juli 2017 am Institut für Ressourcenökologie (IRE) am Helmholtz-Zentrum Dresden-Rossendorf (HZDR) unter der Leitung von Prof. Dr. Thorsten Stumpf und der Betreuung von Dr. Moritz Schmidt in dessen Nachwuchsgruppe „Strukturen und Reaktionen an der Wasser/Mineralgrenzfläche“
durchgeführt.
Ich danke Prof. Dr. Thorsten Stumpf für seine außerordentliche, wissenschaftliche Betreuung, seiner wohlplatzierten, konstruktiven Kritik und den offenen Worten. Ich hatte sehr viel Freude an seinen Bestrebungen mich, durch eine Ausbildung im Fachbereich „Weine aus aller Welt“, dem humboldtschen Bildungsideal einen Schritt näher zu bringen. Und hoffe wir können dies in Zukunft fortsetzen.
Ganz besonderer Dank gilt Herrn Dr. Schmidt für seine exzellente wissenschaftliche Betreuung.
Seine kompetente Unterstützung bei wissenschaftlichen Veröffentlichungen, verwendeten Analysemethoden oder im Hinblick auf meine Doktorarbeit als solches, war stets von unschätzbarem Wert. Auch abseits der Arbeit hatte ich viel Spaß bei Squash, Deep Dish Pizza und vielen unterhaltsamen Abenden.
I am grateful to our cooperation partners in Chicago, Paul Fenter, Sang Soo Lee, Joanne Stubbs, Peter Eng, L. Soderholm, Suntharalingam Skanthakumar. Thank you for your help with sample preparation, measurements, modeling and discussion of the data.
Allen Kollegen des Instituts für Ressourcenökologie danke ich sehr herzlich für die tatkräftige Unterstützung! Ein Spezieller Dank geht an Dr. Vinzenz Brendler für seine Hilfe und dafür, dass er immer ein offenes Ohr hat. Jana Gorzitze danke ich sehr herzlich für ihre administrative Aufopferung! Annette Rumpel und Stephan Weiß danke ich für die tolle Arbeit im Kontrollbereich.
Ich möchte meinen Kollegen für die hervorragende Atmosphäre in unserem schönen Großraumbüro und viele gewinnbringende Diskussionen danken (Ulli, Susi, Constanze, Jérôme, Katharina, Manu, Jackey, Henry). Mein besonderer Dank geht an Sascha, Nina und Carola für die Motivation und das Mitmachen bei sportlichen Aktivitäten oder Kurzweile bei diversen Wohnungsumzügen.
Ich danke meiner Mutter, Elke Hellebrandt, für ihre Unterstützung, ohne die diese Arbeit nicht möglich gewesen wäre.
Meiner Frau Sophia möchte ich für ihre Liebe und Unterstützung danken und dafür, dass sie mir das wertvollste Geschenk überhaupt gemacht hat! Karl danke ich dafür, dass er mich des Nachts zumeist schlafen lässt.
Inhaltsverzeichnis
1 Abstract ... 7
2 Einführung ... 11
2.1 Motivation und Ziele ... 11
2.2 Endlagerung radioaktiver Abfälle ... 13
2.2.1 Das Multi-Barrieren-System ... 13
3 Kenntnisstand ... 15
3.1 Muskovit ... 15
3.2 Sorption - Wechselwirkung von Radionukliden mit Mineraloberflächen ... 17
4 Chemie der Actiniden ... 21
4.1 Allgemeines ... 21
4.2 Aquatische Chemie der untersuchten f-Elemente ... 21
4.2.1 Europium, Americium und Curium ... 23
4.2.2 Leichte Actiniden – Thorium, Uran und Plutonium ... 25
4.2.3 Nanopartikel ... 28
4.3 Grenzflächenreaktionen von Actiniden auf Muskovit ... 30
4.3.1 Sorption von Th an der Basalfläche von Muskovit im Medium NaCl ... 30
4.3.2 Pu(IV)-Nanopartikel aus einer Pu(III)-Lösung. ... 31
4.3.3 Einfluss von Mineraloberflächen auf die Bildung von Pu(IV) ... 32
5 Material & Methoden ... 33
5.1 Probenpräparation ... 33
5.1.1 Allgemeines zur Probenpräparation. ... 33
5.1.2 Oberflächen-Röntgenbeugung. ... 34
5.1.3 Laserfluoreszenzspektroskopie. ... 34
5.1.4 Alpha-Spektrometrie ... 35
5.2 Oberflächen-Röntgenbeugung ... 36
5.2.1 CTR ... 38
5.2.2 RAXR ... 41
5.3 Fluoreszenzspektroskopie ... 44
5.3.1 Allgemeines... 44
5.3.2 Zeitaufgelöste laserinduzierte Fluoreszenzspektroskopie (TRLFS) ... 46
5.3.3 Europium(III)-Fluoreszenz ... 47
5.3.4 Curium(III)-Fluoreszenz ... 50
5.3.5 Verwendete Lasersysteme ... 52
5.4 Weitere Methoden ... 52
5.4.1 GI-XANES ... 52
5.4.2 Spezifische Oberfläche (BET) ... 53
6 Ergebnisse & Diskussion ... 54
6.1 Der Einfluss von Hintergrundelektrolyten auf das Sorptionsverhalten von Thorium an der basalen (001) Fläche von Muskovit ... 54
6.1.1 Thorium-Natriumperchlorat im Vergleich mit Thorium-Natriumchlorid ... 56
6.1.2 Thorium-Natriumperchlorat bei niedrigerer Konzentration (Th-loNaClO4) ... 58
6.1.3 Austausch Experiment NaCl/NaClO4 ... 59
6.1.4 Der Anioneneffekt ... 60
6.1.5 Th-KClO4 System ... 61
6.1.6 Das Lithiumperchlorat-System ... 63
6.1.7 Der Kationeneffekt. ... 65
6.1.8 Mechanismen der Hintergrundelektrolyteffekte ... 67
6.2 Bildung vierwertiger Nanopartikel an der Basalfläche von Muskovit ... 70
6.2.1 Pu(IV)-Nanopartikel ... 70
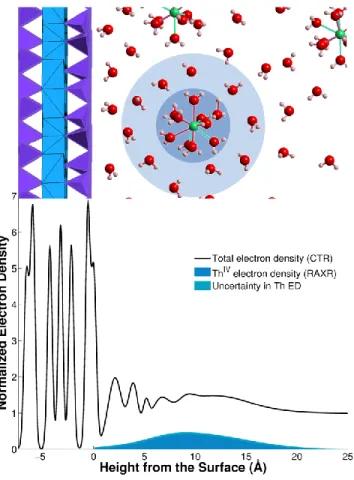
6.2.2 Grenzflächenstruktur - CTR ... 73
6.2.3 RAXR-Daten der Muskovitoberfläche nach der Reaktion mit PuO2 2+ bzw. UO2 2+. ... 75
6.2.4 Unterschiede im Sorptionsverhalten von UO2 2+/PuO2 2+ gegenüber anderen zweiwertigen Ionen. ... 78
6.2.5 Einfluss des Redoxverhaltens ... 79
6.2.6 Bildungsmechanismus von Pu(IV)-Nanopartikeln ... 80
6.2.7 Vergleich der Studie mit anderen Pu-Studien ... 81
6.3 Sorption dreiwertiger f-Elemente an Muskovit und Orthoklas ... 83
6.3.1 Sorption in Abhängigkeit vom pH-Wert ... 83
6.3.2 Sorption von Europium an Muskovit und Orthoklas ... 84
6.3.3 Cm-TRLFS Muskovit und Orthoklas ... 88
6.3.4 Sorption von Am an Muskovit ... 92
6.3.5 IS und Ternärer Orthoklas-Cm-Carbonatkomplex ... 93
6.3.6 Einfluss der Mineralstruktur auf die Sorption ... 94
7 Zusammenfassung ... 95
8 Ausblick ... 98
Abstract
1 Abstract
This thesis investigates the interaction of actinides with siliceous minerals on the molecular level to contribute to a reliable geochemical long-term safety assessment for a nuclear waste disposal site.
The isolation in deep geological formations is the globally preferred strategy for the disposal of actinides like plutonium or americium. The reason is that their high radiotoxicity, due to their high radioactivity and long half-life, requires isolation from the biosphere over a time period up to 1 million years. It is necessary to study the behavior of radionuclides in such a waste disposal site and its surroundings to predict their mobility in the case of a release due to water ingress, and to guarantee that aquifers and the biosphere are not affected. The decisive parameter is the mobility of the radionuclides, which itself depends on different retention mechanisms.
One of these is the sorption on mineral phases. The multi barrier system of a waste disposal site provides a lot of different mineral phases: In the geotechnical barrier are for example swellable clay minerals as bentonite, the geological barrier consists of different mineral phases depending on the host rock (granite, claystone or salt). To improve the understanding of sorption processes, their mechanisms, and what reaction parameters influence them, is the content of the present work. To obtain this understanding on a molecular level, modern analytical techniques must be apllied.
Mainly, Surface X-ray Scattering (SXS), with Crystal Truncation Rod (CTR) and Resonant Anomalous X-ray Reflectivity (RAXR) measurements, and site-selective time-resolved laser fluorescence spectroscopy (TRLFS), were combined with alpha-spectrometry and grazing incidence X-ray absorption near-edge structure (GI-XANES) spectroscopy to obtain and validate results in great detail.
The work can be separated into three major parts: 1. influences of background electrolytes on sorption processes, 2. the influence of the redox chemistry of some actinides on their retention by a mineral surface, and 3. effects of the surface structure on the interaction with adsorbing ions. In the first major part of this work the influence of background electrolytes on the sorption behavior of Thorium ([Th(IV)] = 0.1 mM) at the (001) basal plane of muscovite was investigated. The sorption reaction was investigated by CTR and RAXR measurements in the presence of LiClO4, NaClO4, NaCl and KClO4 (I = 0.1 M or 0.01 M, pH = 3.3 ± 0.3). These measurements directly reveal a strong influence of the background electrolytes on the actinide sorption. No Th sorption was observed in 0.1 M NaClO4, i.e. the surface coverage θ(Th) per unit cell of muscovite (AUC = 46.72 Å2) was ≤ 0.01 Th ions per AUC, in a strong contrast to Th uptake of 0.4 Th/AUC from
a 0.1 M NaCl solution.1 The effect of this anion substitution shows a weak dependence on ionic strength and a limited uptake of 0.04 Th/AUC was detected using 0.01 M NaClO4 as the background electrolyte. Besides this anion effect the electrolyte cation also influences the behavior of Th sorption on muscovite: Weak adsorption was observed in 0.1 M KClO4 (θ(Th) ~ 0.07 Th/AUC), somewhat increased relative to NaClO4 at the same ionic strength. More pronouncedly, using 0.1 M LiClO4 as background electrolyte results in a strong adsorption of θ(Th) = 4.9 Th/AUC. This uptake is ~ 10 times higher than the previously observed uptake in NaCl. The CTR/RAXR structural analysis of the Th- sorption in LiClO4 obtained a broad Th distribution with two distinct peaks centered at 4.1 Å and 29 Å distance from the surface, respectively. Neither the high distribution height of the second species nor the high coverage can be explained by ionic sorption, even if fully hydrated, suggesting that this high uptake is due to the formation and sorption of Th nanoparticles.
All results were qualitatively confirmed by ex situ alpha-spectrometry, where no measurable Th coverage was observed in 0.1 M NaClO4, but 1.6 Th/AUC in 0.1 M LiClO4. The uptake values from alpha-spectrometry is systematically lower for all samples, which can be explained with a washing process that is necessary to measure the samples by alpha-spectrometry, but affects the Th(IV) sorption. In contrast, RAXR measurements are probing the in situ sorption structure.
How the redox behavior of actinides can influence their sorption behavior was investigated at the redox-inactive muscovite (001) surface in contact with solutions containing either 0.1 mM plutonyl(VI) or 1 mM uranyl(VI) (pH = 3.2 ± 0.2 and I(NaCl) = 0.1 M). A combination of in situ X-ray scattering techniques (CTR/RAXR), GI-XANES, and ex situ alpha-spectrometry was used simultaneously to determine the actinide uptake, its sorption structure, and the predominant oxidation state present at the interface. The measured surface coverage is found to be very different for Pu and U. For Pu, alpha-spectrometry found a surface coverage of 8.3 Pu/AUC, which is far in excess of the necessary 0.5 Pu/AUC to compensate the negative surface charge of the muscovite (001) basal plane with PuO2
2+. The adsorbed Pu is broadly distributed (CTR/RAXR) and was identified to be predominantly tetravalent Pu (GI-XANES). Compared to previous findings, these results are consistent with the adsorption of Pu in the form of Pu(IV)-oxo-nanoparticles. In contrast, no adsorbed U was detected at the muscovite interface according to all methods applied, despite the larger concentration of the adsorbate. This difference in the sorption behavior can be explained by the different redox behavior of U and Pu. U will stay mainly in its predominant oxidation state VI, due to its higher stability compared to Pu, while Pu will be present as Pu(VI), but also in a greater extent in its reduced oxidation states. Due to this difference Pu is able to form nanoparticles, whereas this is not the case for U.
Abstract
The third part compares the sorption of trivalent actinides Am(III) and Cm(III) and their homolog Eu(III) on alumino-silicates muscovite (phyllosilicate) and orthoclase (framework silicate). In addition the results from powders from TRLFS single crystals from SXS measurements can be related.
The surface structure shows little influence on the sorption behavior. The amount of sorption plateaus at ~ 100 % at pH > 7 under the chosen conditions ([M] = 10-4 M, [NaCl] = 0.1 M) in both, air and N2. Under acidic conditions all elements adsorb as outer-sphere complexes, while the relative amount of inner-sphere sorption increases with increasing pH. At higher pH a transition from adsorbed aquoions to adsorbed hydrolysis-species and eventually a ternary sorption complex is observed. While TRLFS itself is a strong tool to investigate the speciation of metals in solution or in contact with a mineral surface the complementary use of SXS techniques is able to improve these findings. One question in this context is, however, in how far results from pristine single crystal surfaces are comparable to results from polycrystalline powder samples. In our study no significant differences for the characteristic onset of inner sphere sorption at pH ~ 5 are observed for the sorption of Cm(III) on muscovite powders and Am(III) on muscovite (001).
The results of this work, achieved by using modern methods such as X-ray scattering techniques (CTR/RAXR) and TRLFS, have afforded new insights into the surface reactivity of a wide range of actinides (Th, U, Pu, Am, and Cm) in contact with muscovite and orthoclase. New influence factors could be identified in the influence of the background electrolyte on the sorption behavior, where both cation and anion affect the actinide´s retention. The different redox behavior of redox-active actinides like U and Pu also influenced the sorption behavior tremendously, even in contact with the redox-inactive mineral muscovite. The complementary use of surface X-ray diffraction and laser spectroscopic methods was instrumental to enable us to investigate the distribution, occupancy, and speciation of the radionuclides at mineral surfaces on a molecular level, which will be required to make a reliable safety assessment of any potential nuclear waste disposal site.
Einführung
2 Einführung
2.1 Motivation und Ziele
Ziel dieser Arbeit ist es, die Wechselwirkung endlagerrelevanter Actiniden mit Mineralen zu untersuchen und grundlegende Kenntnisse über Prozesse an der Wasser/Mineralgrenzfläche zu erlangen. Dabei sollten Mechanismen der Sorption von Actiniden und damit assoziierte Reaktionen wie bspw. die Bildung von Nanopartikeln aufgeklärt werden. Im Fokus stand dabei der Erkenntnisgewinn auf molekularer Ebene, welcher durch den Einsatz der Oberflächen- Röntgenbeugung (SXS) und Laserfluoreszenzspektroskopie ermöglicht wurde. Die korrekte Beschreibung der zu erwartenden Reaktionen kann dann Grundlage für eine thermodynamische Parametrisierung und schließlich Ausbreitungsrechnungen als Beitrag zum safety case eines Endlagerstandorts liefern.
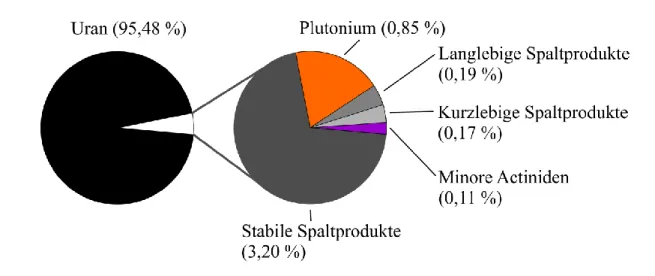
In vielen industriellen und medizinischen Bereichen kommen radioaktive Stoffe zum Einsatz, insbesondere bei der Energieerzeugung in Kernkraftwerken. Die dabei anfallenden Abfälle sollen in Deutschland endgelagert werden. Im Mittelpunkt der Untersuchungen der vorliegenden Studie standen dabei jene Elemente, die zum Inventar abgebrannter Kernbrennelemente (Brennstäbe) gehören und deren Radiotoxizität über lange Zeiträume bestimmen. Die Endlagerung dieser hochaktiven, wärmeentwickelnden Abfälle stellt dabei neben der technischen auch eine wissenschaftliche Herausforderung dar. Grund dafür ist die Zusammensetzung und die daraus resultierenden Eigenschaften dieses Abfalls (Abbildung 1). Er besteht hauptsächlich aus Uran (95 %), vermischt mit stabilen und instabilen Spaltprodukten, Plutonium und den sogenannten Minoren Actiniden (Neptunium, Americium, Curium). Dabei wird die Radiotoxizität (Abbildung 2) anfänglich von den Spaltprodukten und deren Zerfallsprodukten und nach etwa 100 Jahren, nach der Entnahme aus dem Reaktor, durch Pu und die Minoren Actiniden, v.a. Am, dominiert.
Aufgrund der relativ kurzen Halbwertszeiten der Spaltprodukte klingt deren Radiotoxizität schon nach wenigen hundert Jahren unter das Referenzniveau der Radiotoxizität von Uranerz ab. Erst nach einer relativ langen Zeit von etwa 10000 Jahren ist die Radiotoxizität der Minoren Actiniden und deren Zerfallsprodukte auf diesem Niveau angekommen. Obwohl Plutonium nur einen sehr geringen Anteil von etwa 0,85 % der Gesamtmasse eines abgebrannten UO2-Brennstabs ausmacht, dominiert dessen Radiotoxizität über den längsten Zeitraum und erreicht das Referenzniveau erst nach mehr als 100000 Jahren. Dies erklärt die Notwendigkeit einer langfristigen, sicheren Endlagerung hochradioaktiven Abfalls und den Fokus dieser Arbeit auf diese Elemente, um eine Gefährdung der Umwelt und des Menschen durch eine Kontamination mit diesen Stoffen auszuschließen. Für die adäquate Einschätzung der Sicherheit einer solchen Endlagerung ist es notwendig zu verstehen, wie sich gerade diese Radionuklide im Nah- und Fernfeld eines Endlagers
verhalten. Dies wiederum bedarf des Prozessverständnisses dieses Verhaltens auf molekularer Ebene.
Abbildung 1: Zusammensetzung eines verbrauchten Kernbrennelements nach der Verwendung in einem Druckwasserreaktor (PWR 33 GW/t, 10 Jahre gekühlt)2
Abbildung 2: Radiotoxizität abgebrannter Brennelemente im Vergleich zur Referenz Uranerz2
Einführung
2.2 Endlagerung radioaktiver Abfälle
Radioaktive Abfälle werden in nicht wärmeentwickelnde (schwach- bis mittelaktive) und wärmeentwickelnde (hochaktive) Abfälle unterschieden. Erstere werden verbrannt oder komprimiert und in ein tiefengeologisches Endlager eingebracht. Die technischen Anforderungen an ein solches Endlager sind geringer, als dies der Fall für ein Endlager für hochradioaktive Abfälle ist. In Deutschland ist für schwach- bis mittelaktive Abfälle der Schacht Konrad vorgesehen. Eine Entscheidung über den Standort des Endlagers für hochaktive wärmeentwickelnde Abfälle steht noch aus und soll durch das am 23.07.2013 verabschiedete Standortauswahlgesetz (StandAG) geregelt werden. Der Gesetzgeber sieht vor, dass die sichere Endlagerung für einen Zeitraum von 1 Million Jahren gewährleistet werden muss. Für diesen geologisch betrachtet kurzen Zeitraum kann die großräumige tektonische Entwicklung vorausgesagt werden. Wird dieser Zeitraum allerdings an der Menschheitsgeschichte gemessen, so ist er gleichsam lang und eine Abschätzung erweist sich als schwierig. Laborexperimente können einen solchen Zeitraum nicht abbilden.
2.2.1 Das Multi-Barrieren-System
Das Konzept der tiefengeologischen Endlagerung mithilfe eines Multi-Barrieren-Systems entspricht dem aktuellen internationalen Konsens. Die innerste Barriere stellt die Abfallform selbst dar. Drei weitere Barrieren umschließen diese in folgender Reihenfolge: Die technische, die geotechnische sowie die geologische Barriere. Der Behälter in dem der Abfall (unabhängig seiner Form) in das Endlager eingebracht wird, befindet sich im direkten Kontakt mit der entsprechenden Abfallform und kann je nach Wirtsgestein aus verschiedenen Materialen bestehen, z. B. Stahl oder Kupfer. Dem folgt die geotechnische Barriere, welche abhängig vom Wirtsgestein z. B. aus quellfähigen Materialen (in Granit- und Tongesteinen i. d. R. Bentonit) mit guten Sorptionseigenschaften bestehen kann. Zum einen soll diese Barriere als Füllmaterial dienen und einen Wassereintritt in das Endlager verhindern, zum anderen soll sie im Falle eines Wassereintritts eine möglichst effektive Rückhaltung eventuell gelöster Radionuklide durch ihre sehr guten Sorptionseigenschaften gewährleisten. Sollten diese Barrieren überwunden werden, agiert das Wirtsgestein, in dem sich das Endlager befindet, als letzte Barriere, um eine Kontamination von Aquiferen und der Biosphäre zu verhindern. Mit der Wahl des Wirtsgesteins gehen bestimmte petrographische Eigenschaften einher, was die spezifische Ausgestaltung der technischen und geotechnischen Barriere beeinflussen kann. So stehen in Deutschland Tonformationen, Steinsalz und Granit zur Debatte, wobei jedes dieser Wirtsgesteine Vor- und Nachteile besitzt.
Tonformationen besitzen, bei einem ausreichend hohen Anteil quellfähiger Tonminerale der Smektit-Gruppe, hervorragende „Selbstheilungseigenschaften“, im Falle tektonisch oder anthropogen induzierter Bildung von Störungen, z. B. durch den Bau des Endlagers selbst hervorgerufene Störungen. Zudem besitzen Tonminerale hervorragende Sorptionseigenschaften und eine geringe Wasserleitfähigkeit, beides trägt zu einer verringerten Mobilität möglicher Kontaminanten bei. Ingenieurstechnisch ist der Bau eines Endlagers in einer Tonformation allerdings herausfordernd. Da durch den radioaktiven Zerfall der hochaktiven Abfälle im Nahfeld eines Endlagers hohe Temperaturen auftreten können, ist auch die schlechte Wärmeleitfähigkeit von Tongestein nachteilig. Die Wärmleitfähigkeit von Steinsalz ist dagegen hervorragend und auch die technische Umsetzung des Baus eines Endlagers ist deutlich einfacher. Aufgrund der sowohl viskosen als auch plastischen Eigenschaften von Salz können mögliche Hohlräume ebenfalls effektiv geschlossen werden. Die hohe Löslichkeit des Steinsalzes ist hingegen von Nachteil, da ein möglicher Wassereintritt nur minimal oder gar nicht verhindert wird. Bei trockenen Bedingungen findet im Salz allerdings kein Transport statt. Der einfache und sehr wahrscheinliche Wasserzutritt stellt auch die größten Probleme für ein Endlager im Wirtsgestein Granit dar. Granit ist ein sehr stabiles aber sprödes Material, welches viele Störungen, d. h. eine starke Zerklüftung, aufweist.
Diese Klüfte können potentiell große Mengen Wasser mit hohen Fließgeschwindigkeiten führen.
Der Bau des Endlagers sollte sich in Granit ingenieurstechnisch allerdings als am Einfachsten erweisen und die Limitierungen der geologischen Barriere können teilweise durch Änderungen in der technischen (Bspw. durch Verwendung von Cu-Behältern, wie in Schweden oder Finnland vorgesehen) sowie der geotechnischen Barriere ausgeglichen werden.
Die Wahl des in dieser Arbeit hauptsächlich untersuchten Minerals, Muskovit, begründet sich auf dessen Relevanz im Konzept des Multi-Barrieren-Systems. Zum einen ist Muskovit aufgrund der ähnlichen Strukturen ein hervorragendes Analogon für Tonminerale, aus denen Tongesteine und Bentonit (geotechnische Barriere) zu großen Teilen bestehen. Zum anderen sind Glimmer, zu denen Muskovit gezählt wird, eines der gesteinsbildenden Minerale von Granit. Die Relevanz von Muskovit wird im Detail in Kapitel 3.1 erörtert.
Kenntnisstand
3 Kenntnisstand
3.1 Muskovit
In der vorliegenden Arbeit wurde das Sorptionsverhalten an Muskovit, einem Glimmervertreter, untersucht. Muskovit tritt ubiquitär in der Natur auf und kann in Form von Einkristallen im Zentimeter- bis Metermaßstab gesteinsbildend auftreten. Häufiger sind allerdings kleinere Kristalle, welche sich z. B. in Sandsteinen porenständig als Kutane um die Körner gebildet haben. Dadurch ist das metamorph entstandene Mineral im direkten Kontakt zu potentiell grundwasserführenden Porenräumen. Im Falle einer Grundwasserkontamination durch Radionuklide, können hier also entsprechend wirkende Rückhaltemechanismen direkt greifen.
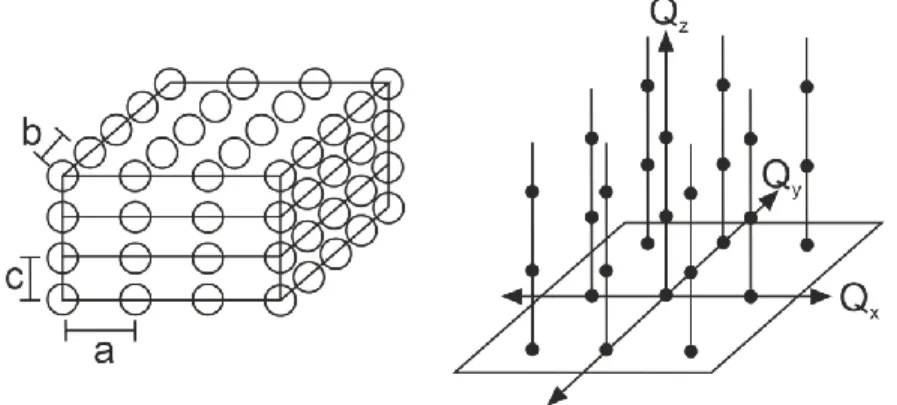
Muskovit KAl2(AlSi3O10)(OH,F)2 ist ein dioktaedrisches Schichtsilikat (Phyllosilikat) mit einer Tetraeder-Oktaeder-Tetraeder (TOT) Schichtstruktur (Abbildung 3). Drei Sauerstoffe eines SiO4- Tetraeders werden mit angrenzenden Tetraedern geteilt, wodurch diese in einer Ebene miteinander verbunden sind. Das verbleibende Sauerstoffion verbindet die Schichten der Tetraeder mit denen der AlO6-Oktaeder, welche sich zwischen den zwei Tetraederschichten befinden. Die zwei Tetraederschichten sind dabei um 180° zueinander rotiert.3,4
Abbildung 3: (links) Die Polygondarstellung zeigt die Struktur von Muskovit mit Blickrichtung entlang der Raumrichtung a. Die SiO4-Tetraeder (blau) sind durch die Sauerstoffe an den Spitzen mit den AlO6-Oktaedern (Orange) verbunden, wobei die TOT Schichten durch K+-Ionen (rot) in den Zwischenschichten miteinander verbunden sind. (rechts) Natürlicher Muskovit vergesellschaftet mit Apatit Kristallen.
Die hier verwendete Modifikation Muskovit-2M1ist monoklin und kann große Einkristalle ausbilden. Durch die Substitution von vierwertigem Si durch dreiwertiges Al besitzt jede TOT- Schicht eine permanente negative Strukturladung von 1e- pro Einheitszelle, deren Fläche
AUC = 46,72 Å2 beträgt, dies entspricht 0,021 e-/Å2. Diese Ladung wird im Kristallbulk („Körper“
des Kristalls) durch K+-Ionen kompensiert, die in den Zwischenschichten sitzen. Spaltet man den Kristall entlang seiner (001) Fläche und bringt diese Spaltfläche in Kontakt mit Wasser, werden die K+-Ionen entfernt und die so entstandene Oberflächenladung wird durch Ionen aus der Lösung kompensiert.5-7 Entlang dieser (001) Fläche besitzt Muskovit eine vollkommene Spaltbarkeit, diese führt zu annähernd atomar-flachen Oberflächen.
Ein Großteil der Untersuchungen dieser Arbeit wurde an der Basalfläche von Muskovit durchgeführt. Diese Fläche ist aufgrund der konstanten permanenten Oberflächenladung sehr gut für Sorptionsexperimente verschiedener Art geeignet. Desweiteren ist es durch die atomar-flachen Oberflächen deutlich einfacher möglich Mechanismen aufzuklären, ohne Unsicherheiten durch ein mögliches Zusammenspiel unbekannter oder nicht klar definierter Oberflächensites zu generieren.
Die Ergebnisse an Muskovit in Pulverform, wie er für die Laserfluoreszenzmessungen genutzt wurde, integrieren über vorhandene Spezies an allen möglichen Sites, z. B. an Kanten oder Ecken des Minerals. Dies macht eine eindeutige Identifizierung der Lokalität z. B. einer Sorptionsspezies schwierig, es spiegelt aber das natürliche System besser wieder. Die Vergleichbarkeit beider Ansätze wird im Kapitel 6.3 anhand der Sorption von An(III) näher betrachtet.
Relevanz als Wirtsgestein für Endlager
Granit und Gneis bestehen hauptsächlich aus Quarz, Feldspat und Glimmer. In diesen Gesteinen nehmen Glimmer eine besondere Position hinsichtlich deren Rückhaltevermögen ein. Die Sorption von Radionukliden ist an Muskovit stärker ausgeprägt als an Quarz oder Feldspat.8,9 Demzufolge ist bei der Entscheidung für den Bau eines Endlagers in Granit, wie dies z. B. im finnischen Olkiluoto der Fall ist, der gesteinsbildende Glimmeranteil von entsprechender Relevanz für eine potentielle Rückhaltung von Radionukliden.
Muskovit kann ebenso als effektives Analogon für viele Tonminerale verwendet werden, da diese ähnliche Kristallgitter ausbilden. Ein Beispiel ist Montmorillonit, ein Tonmineral der Smektitgruppe. Genau wie Muskovit sind auch Smektite durch eine typische TOT-Gitterstruktur im monoklinen Kristallsystem aufgebaut und besitzen eine permanente Oberflächenladung aufgrund der Substitution von Si4+ gegen Al3+ im Kristallgitter. Entsprechend sind die oberflächenaktiven Gruppen identisch und es ist anzunehmen, dass oberflächenladungsinduzierte Prozesse, wie z. B.
Sorption von innersphärischen und äußersphärischen Komplexen, ähnlich funktionieren. Hier sei allerdings zu erwähnen, dass die Oberflächenladung von Montmorillonit mit 0,6 e-/AUC deutlich geringer ist als die von Muskovit mit 1e-/AUC. Der wesentliche Unterschied ist die Fähigkeit der Smektite die Kationen ihrer Zwischenschichten zu substituieren, wodurch Smektite quellfähig sind.
Kenntnisstand
Trotzdessen eröffnet die Analogie zu Muskovit die Möglichkeit, Systeme mit Methoden zu untersuchen, die aufgrund der Morphologie des Montmorillonit nicht direkt an diesem Mineral möglich sind. Ein Beispiel dafür ist die in dieser Arbeit angewandte SXS, für deren Durchführung ausreichend große Einkristalle notwendig sind.
Relevant wird der Zusammenhang Muskovit-Smektit auch durch die potentielle Verwendung von Bentonit als Füllmaterial der geotechnischen Barriere (siehe Kapitel 2.2.1) eines Endlagers für hochradioaktive Abfälle. Denn Bentonit beinhaltet neben geringen Anteilen an Muskovit selbst meist mehr als 70 % Smektit.10 Daher können Forschungsergebnisse, die an Muskovit erzielt wurden, analog für das Material der geotechnischen Barriere und für Tonformationen als Wirtsgestein Verwendung finden.
Orthoklas
Der Fokus dieser Arbeit liegt in der Analyse des Sorptionsverhaltens von endlagerrelevanten Radionukliden an dem Mineral Muskovit (KAl2(AlSi3O10)(OH,F)2). Zusätzlich dient Orthoklas als Vergleichsmineral mit ähnlichen Eigenschaften und wurde parallel mit Hilfe von Laserfluoreszenzmessungen untersucht. Orthoklas (KAlSi3O8) ist das K-Endglied der Mischungsreihe der Alkali-Feldspäte. Die Gruppe der Feldspäte kommt in der Natur gleichsam dem Muskovit ubiquitär vor und stellt den Großteil der gesteinsbildenden Minerale der Erdkruste. Das monokline Gerüstsilikat ist aus Al- und Si-Tetraedern aufgebaut, welche über O-Atome verbrückt sind. Aufgrund der Substitution von Si4+ durch Al3+ ist das Gerüst negativ geladen. Dieser Austausch in einem von vier Tetraedern führt dazu, dass der Hohlraum zwischen diesen Tetraedern durch ein Kaliumion besetzt ist,11 wodurch die Ladung im Kristallgitter kompensiert wird. Das Kaliumion ist neunfach koordiniert. Orthoklas besitzt vollkommene Spaltbarkeiten entlang der (001) und (010) Flächen. Das Mineral soll in dieser Arbeit der Identifizierung dienen, inwiefern die chemisch sehr ähnlichen Minerale Orthoklas und Muskovit aufgrund struktureller Unterschiede, Unterschiede in der Wechselwirkung mit Radionukliden aufweisen.
3.2 Sorption - Wechselwirkung von Radionukliden mit Mineraloberflächen
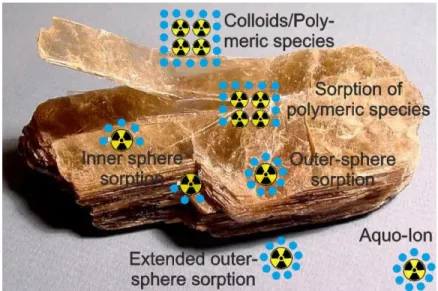
Gegenstand der vorliegenden Arbeit sind Sorptionsprozesse von Actiniden und Lanthaniden an der Fest-Flüssig-Grenzfläche von negativ geladenen Mineralen und der Lösung mit der diese in Kontakt stehen. Unter Sorption sind elektrostatische Wechselwirkungen von Ionen mit Mineraloberflächen zusammengefasst. Je nach Art der wirkenden Kräfte wird zwischen Physi- und Chemisorption unterschieden. Bei der Physisorption treten relativ schwache elektrostatische Bindungen auf. Abhängig von der Anzahl intakter Hydrathüllen aus Wassermolekülen eines Ions
und dessen Abstand zur Oberfläche, wird zwischen außersphärischen bzw. outer sphere Komplexen (OS-Komplexe) und erweitert-außersphärischen bzw. extended outer sphere Komplexen (E-OS- Komplexe) unterschieden (siehe Abbildung 4). Von innersphärischen oder inner sphere Komplexen (IS-Komplexe) ist die Rede, wenn Teile der Hydrathülle des Sorbats durch Komplexierung mit Oberflächengruppen des Substrats ersetzt worden sind. Diese Art der Sorption wird auch als Chemisorption bezeichnet, da nun eine chemische Bindung zur Oberfläche besteht. Neben der Sorption von Ionen kann es zudem zur Sorption von Polymeren oder Nanopartikeln kommen. Diese können direkt an der Oberfläche oder in Lösung entstehen. Eine weitere Form der Sorption ist der Einbau von Ionen in das Wirtskristallgitter. Dieser kann unter bestimmten Bedingungen (Wachstum oder Umkristallisation) innerhalb geeigneter Minerale, bspw. bei guter Übereinstimmung der Ionenradien von Wirts- und Gastion, stattfinden.
Abbildung 4: Adsorptionsprozesse von Radionukliden mit Mineraloberflächen.
Es ist notwendig Sorptionsprozesse zu studieren, denn diese stellen einen der wichtigsten Einflüsse auf das Migrationsverhalten von Kontaminanten, z. B. Radionukliden, und damit einen der wichtigsten Rückhaltemechanismen in der Umwelt dar. Dabei ist die Sorption ein komplexer Vorgang, welcher durch eine Vielzahl von Faktoren beeinflusst wird. Einer der wichtigsten Faktoren ist der pH-Wert, welcher z. B. die Speziation in Lösung oder die Ladung von Mineraloberflächen beeinflussen kann. Durch Variation des pH-Wertes kann das Zeta-Potential von Partikeln in Suspension bestimmt werden,12 dies lässt Rückschlüsse auf dessen Oberflächenladung zu. Die Oberflächenladung von Muskovit und Orthoklas in Pulverform ist im gesamten
Kenntnisstand
Untersuchungsbereich dieser Arbeit negativ und erreicht den isoelektrischen Punkt9 nicht. Die Oberflächenladung der (001) Fläche des Muskovits hingegen ist vom pH-Wert unabhängig, da diese permanente Ladung durch Substitution unterschiedlich geladener Ionen im Kristallgitter hervorgerufen wird.
Ein weiterer Einflussfaktor auf die Sorption eines bestimmten Ions ist die Konkurrenz mit anderen gelösten Ionen, z. B. dem Hintergrundelektrolyt oder dem Lösungsmittel, Wasser, selbst.
Die ein bis zwei adsorbierten Wasserschichten können dabei je nach Oberfläche relativ stark gebunden sein13 und sind in der Regel innerhalb der ersten 3 Å über der Oberfläche lokalisiert.14 Schon diese Wasserschichten stellen für die Sorption von Radionukliden eine Barriere dar.
Bezüglich des Hintergrundelektrolyts spielt nicht nur die Ionenstärke sondern auch die Wahl des Hintergrundelektrolyts eine Rolle, obwohl dieses lediglich die Ionenstärke eines Systems konstant halten sollte. Denn die unterschiedlichen Kationen und Anionen des Hintergrundelektrolyts konkurrieren unterschiedlich stark mit unterschiedlichen Radionukliden und beeinflussen die Sorption in ihrer Art und Weise und Oberflächenbelegung dadurch unter Umständen entscheidend (siehe Kapitel 6.1). Eine Reihe weiterer Parameter können das Sorptionsverhalten von Ionen an einem Mineral beeinflussen: unter anderem Temperatur, Eh-Wert, Druck, Korngröße des Minerals, wobei letzteres die Oberflächensites und deren Dichte beeinflussen kann.
Als Desportion wird der Umkehrprozess der Sorption bezeichnet. Ob und wie leicht ein gebundener Stoff freigesetzt werden kann, hängt dabei vom Typ der Sorption ab. Schon Änderungen der Ionenstärke oder der Fließgeschwindigkeit des Grundwassers kann zur Desorption von außersphärischen Sorptionskomplexen führen. Um innersphärische Sorptionskomplexe zu desorbieren, ist i. A. eine Änderung des pH-Wertes notwendig. Zur Freisetzung einer Einbauspezies hingegen ist es i. d. R. notwendig das Wirtsmineral aufzulösen.
Häufig wird Sorption nur quantitativ betrachtet, das heißt, es werden Sorptionsisothermen anhand der Konzentration eines sorbierten Ions oder bei unterschiedlichem pH-Wert ermittelt, um daraus den Verteilungskoeffizienten KD zu berechnen. Dieser ist definiert als Konzentrationsverhältnis eines Stoffes zwischen zwei nicht mischbaren Phasen.15 Diese Werte sind spezifisch für bestimmte Lösungsbedingungen und enthalten keine mechanistischen Informationen, weshalb sich ein Vergleich verschiedener Systeme häufig als schwierig erweist. Um Aussagen über verschiedene Sorptionsspezies treffen zu können, müssen daher Methoden eingesetzt werden, welche in der Lage sind, die einzelnen Spezies zu unterscheiden. Die Identifizierung der Sorptionsprozesse und der zugrunde liegenden Reaktionsmechanismen wurde in dieser Arbeit durch die Verwendung der Methoden Oberflächen-Röntgenbeugung (SXS) und zeitaufgelöster
laserinduzierte Fluoreszenzspektroskopie (TRLFS) möglich (siehe Kapitel 5.2 & 5.3). Ohne chemisches Verständnis sind Extrapolationen zum Verhalten und der Verteilung von Radionukliden in der Umwelt nur unzureichend möglich.
Chemie der Actiniden
4 Chemie der Actiniden
4.1 Allgemeines
Als Actinide (An) werden die Elemente mit den Ordnungszahlen von 90 bis 103 bezeichnet, die dem Actinium folgen und die 5f-Elektronenschale auffüllen. Eine Besonderheit der Actiniden ist das Fehlen von stabilen Nukliden, alle Isotope dieser Elemente sind radioaktiv. In den Experimenten dieser Arbeit wurden Thorium (90), Uran (92), Plutonium (94), Americium (95) und Curium (96) verwendet.
Einige Actinidisotope kommen als primordiale Nuklide (235U, 238U und 232Th) vor, d. h. diese Nuklide sind aufgrund ihrer langen Halbwertszeiten (> 108 a) Bestandteile der Erde seit ihrer Entstehung und bis heute nicht vollständig zerfallen. Die Elemente mit einer höheren Ordnungszahl als Uran, die sogenannten Transurane kommen nicht mehr in der Natur vor, sondern entstehen bspw. durch Neutroneneinfang, bei hohen Neutronenflüssen in einem Leistungsreaktor. Dabei entsteht, wie der Hauptreaktionspfad in Gleichung 4.1 zeigt, initial 239U, welches sich daraufhin in zwei aufeinander folgenden β--Zerfällen zu 239Pu umwandeln kann. In Folgereaktionen wie im Beispiel von Gleichung 4.2 gezeigt ist, können dann auch die Nuklide des Am und Cm entstehen.
(4.1)
(4.2)
Abbildung 1 zeigt die Zusammensetzung eines abgebrannten Brennelements nach der Entnahme aus dem Reaktor. Hieraus wird deutlich, dass die gebildete Menge an Americium und Curium relativ gering ist. Aufgrund dessen werden diese Elemente, zusammen mit Neptunium als Minore Actinide bezeichnet. Das Interesse an diesen Elementen, entsteht somit nicht aus ihrer Quantität, sondern aus der besonders hohen Radiotoxizität, die über viele tausend Jahre anhält (Abbildung 2).
4.2 Aquatische Chemie der untersuchten f-Elemente
Die Actiniden können in „leichte“ Actiniden (Th bis Pu) und „schwere“ Actiniden (ab Am) eingeteilt werden. Diese Einteilung orientiert sich neben der Ordnungszahl zudem an den stabilen Oxidationsstufen in wässriger Lösung (Tabelle 1). Die leichten Actiniden besitzen eine Vielzahl stabiler Oxidationsstufen in wässriger Lösung, wohingegen die schweren Actiniden in wässriger Lösung i. d. R. in ihrer dreiwertigen Oxidationsstufe vorliegen. Der Grund für dieses
unterschiedliche Verhalten ist die Delokalisierung der 5f-Elektronen im Falle der leichten Actiniden, dadurch überlagert sich das 5f- mit dem 6d-Orbital. Im Falle der schwereren Actiniden sind die 5f-Elektronen lokalisiert, wodurch diese Valenzelektronen separiert sind und nur diese für chemische Reaktionen zur Verfügung stehen.
Die Chemie der meisten Lanthanide ähnelt jener der schweren Actiniden (siehe Kapitel 4.2.1), da in wässriger Lösung ebenfalls die Oxidationsstufe III dominiert. Die Ionenradien dreiwertiger Lanthaniden ähneln denen der dreiwertigen Actiniden ebenfalls. Aufgrund dieser Gemeinsamkeiten können dreiwertige Lanthaniden sehr gut als chemische Analoga für die dreiwertigen Minoren Actiniden Pu, Am und Cm verwendet werden. Lanthanide füllen die 4f-Schale und Actinide die 5f- Schale mit Elektronen. Die Lanthanide und Actinide werden auch unter dem Begriff f-Elemente zusammengefasst. Im Gegensatz zur 4f-Schale der Lanthanide ist die 5f-Schale der Actinide schwächer abgeschirmt, daraus resultieren unter anderem deutlich höhere kovalente Bindungsanteile bei den Actiniden. 16,17
Die möglichen Oxidationsstufen der Lanthaniden und Actiniden sind in Tabelle 1 dargestellt.17
Tabelle 1: Auftretende Oxidationsstufen der Lanthanide und der Actinide (in Lösung)*, fett dargestellt sind die stabilsten Oxidationsstufen und die Namen der Elemente die Gegenstand dieser Arbeit sind
La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
2 2 2 2
3 3 3 3 3 3 3 3 3 3 3 3 3 3 3
4 4 4 4 4
Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr
2 2 2
3 3 3 3 3 3 3 3 3 3 3 3 3
4 4 4 4 4 4 4 4
5 5 5 5 5
6 6 6 6 6
7 7
*nach Holleman & Wiberg 2007
Chemie der Actiniden
4.2.1 Europium, Americium und Curium
Unter den Seltenerdmetallen ist das Europium tatsächlich selten, nach Promethium ist es das zweitseltenste Element dieser Gruppe. Die Elektronenkonfiguration von Eu ist [Xe]4f76s2 und die des Eu3+ [Xe]4f6. Americium ist ein radioaktives Element aus der Gruppe der Actiniden. Die Elektronenkonfiguration von Americium ist [Rn]5f77s2. Am3+ besitzt dementsprechend die Elektronenkonfiguration [Rn]5f6. In dieser Arbeit wurde das radioaktive Nuklid 243Am verwendet, welches ein Alpha-Strahler mit einer Halbwertszeit von 7400 Jahren ist. Curium ist ebenfalls ein radioaktives Element der Gruppe der Actiniden und folgt dem Americium im Periodensystem. Die Elektronenkonfiguration ist [Rn]5f76d17s2, die von Cm3+ [Rn]5f7. Der Ionenradius von Cm3+ beträgt 0,97 Å (Koordinationszahl 6).18 In dieser Arbeit wurde das radioaktive Nuklid 248Cm verwendet, welches ein Alpha-Strahler mit einer Halbwertszeit von 3,48 × 105 Jahren ist. Aus strahlenschutztechnischen Gründen wurden für die Experimente dieser Arbeit die erwähnten langlebigen Nuklide verwendet (243Am, 248Cm).
Dreiwertiges Europium wird aufgrund der erwähnten ähnlichen Eigenschaften in dieser Arbeit als Anaolog für dreiwertiges Americium und Curium verwendet. Die dreiwertigen Ionen dieser Metalle agieren, aufgrund der niedrigen Ionenradien von ~ 100 pm18 und der daraus resultierenden hohen Ladungsdichte, als harte Lewis-Säuren nach dem Prinzip der harten und weichen Lewissäuren (hard and soft acids and bases, kurz HSAB nach Pearson19). Als solche reagieren sie bevorzugt mit harten Lewisbasen, wie beispielsweise OH-, CO3
2- oder Cl-. Eine wichtige Reaktion in der aquatischen Chemie harter Lewissäuren ist somit die Hydrolyse (Gleichung 4.3a-c).20 Dabei werden mit steigendem pH-Wert die verschiedenen Hydrolysespezies gebildet und schon bei neutralem bis schwach basischem pH-Wert fällt das in Gleichung 4.3c dargestellte Produkt Eu(OH)3, bei [Eu3+] = 10-4 M, aus. Die Hydrolysekonstanten der dreiwertigen Oxidationsstufe spiegeln die Ähnlichkeit wieder: Cm(OH)2+ log0β11 = -6,8 ± 0,521,22 und Am(OH)2+
log0β11 = -7,2 ± 0,523 im Vergleich zu Eu(OH)2+ log0β11 = -7,76.24
(4.3a) (4.3b) (4.3c)
Die Speziation mit und ohne den Einfluss von Carbonat (CO3
2-) am Beispiel von Europium ist in Abbildung 5 dargestellt. Die Berechnungen wurden mithilfe des PHREEQC Lösungs-
Modellierungspakets (Version 2.18) anhand von Daten der NAGRA/PSI chemisch- thermodynamischen Datenbank durchgeführt.25
Abbildung 5: Speziationsberechnung unter atmosphärischen (links) und inerten (rechts) Bedingungen, [Eu] = 1 × 10-4 M, [NaCl] = 0,1 M.
Es sind geringe Unterschiede zwischen atmosphärischen und inerten Bedingungen zu verzeichnen. Für die Sorption an den Mineraloberflächen zur Verfügung stehende Komplexe sind ab ca. pH 6,5 die erste Hydrolysespezies bzw. unter Atmosphäre auch der erste Carbonatokomplex.
Bei pH ~ 7,5 ist zudem mit wenigen Prozent die zweite Hydrolysespezies in Lösung vorhanden. Für die Feststellung, welche dieser Spezies am Sorptionsprozess beteiligt sind, werden im Kapitel 6.3 TRLFS-Untersuchungen mit Europium und mit Curium dargestellt.
Unter sauren Bedingungen liegen die Metallkationen als Aquoion M3+ vor und sind 9-fach koordiniert.26 Die Stabilität von Komplexen mit einigen Liganden richtet sich nach folgender Sequenz: CO3
2- > OH- > F- > H2PO4 > NO3
- > Cl- > ClO4
-. Generell ist die Hydrolyse in wässrigen Lösungen stark favorisiert, wobei die Komplexierung mit Hydroxiden bei pH > 5 beginnt. Dabei werden die Komplexe entsprechend der generellen Formel geformt, wobei n = 1-3 sein kann (Gleichung 4.3a-c). M-Carbonato-Komplexe werden nach der generellen Formel , wobei n = 1-3 annehmen kann, gebildet.
Von besonderem Forschungsinteresse ist Am, da es einen relevanten Anteil der Aktivität abgebrannter Kernbrennelemente ausmacht. Während die Aktivität von Pu (nach 30 Jahren Abklingen) anfänglich dominiert, übernimmt Am diese Rolle ab etwa 300 Jahren (nach der Entladung des Reaktors) um nach etwa 10 000 Jahren vollständig abgeklungen zu sein und erneut von Pu mit dem höchsten Aktivitätsanteil abgelöst zu werden.27
Chemie der Actiniden
4.2.2 Leichte Actiniden – Thorium, Uran und Plutonium
Thorium besitzt die Elektronenkonfiguration [Rn]6d27s2, es besitzt also keine 5f-Elektronen. In wässriger Lösung liegt Thorium ausschließlich in seiner vierwertigen Oxidationsstufe vor, die aufgrund der [Rn]-Elektronenkonfiguration enorm stabil ist. Dies ermöglicht die Untersuchung von Sorptionsprozessen unabhängig von Redoxprozessen. Das größte Ion der vierwertigen Actiniden, Th4+ ist gleichzeitig das weichste und basischste der vierwertigen Kationen. Die Hydrolyse von Th(IV) ist schon bei stark sauren pH-Werten sehr ausgeprägt und dominiert die Speziation durchweg. Die Speziation verhält sich dabei für 0,1 M NaCl bei niedrigen Th-Konzentrationen ([Th(IV) = 10-8 M)28,29 folgendermaßen: Die ersten drei Hydrolysespezies [Th(OH)]3+/[Th(OH)2]2+/[Th(OH)3]+ haben ihre Maxima bei pH = 3,8/4,8/5,7 und ab pH = 6,1 dominiert Th(OH)4. Bei steigender Th-Konzentration verschiebt sich diese Speziation aufgrund der Bildung von Th-Polymeren deutlich. Schon bei pH > 1 bilden sich Dimere mit unterschiedlichen Verbrückungen und Koordinationszahlen.30,31 Die bereitwillige Bildung von Th- Polymeren/Nanopartikeln32,33 beginnt teilweise schon bei niedrigen pH-Werten (ab ~ pH 1,5). Diese können sowohl amorpher als auch mikrokristalliner Natur sein.34
Das verwendete Nuklid von Thorium ist der radioaktive primordiale Alpha-Strahler 232Th mit einer Halbwertszeit von 1,4 × 1010Jahren.
Plutonium. Die Elektronenkonfiguration von Pu ist [Rn]5f67s2 und die Oxidationsstufen III bis VI können in wässrigen Lösungen vorliegen. Grund dafür sind die sehr ähnlichen elektrochemischen Potentiale der Redoxpaare von Pu unter sauren Bedingungen von etwa 1 V (z.B.
PuVI/PuV = 0,913; PuVI/PuIV = 1,043; PuV/PuIV = 1,170; für 298,15 K in V vs. SHE (Standardwasserstoffelektrode) in 1 M HClO4).35 Abbildung 6 zeigt, dass PuO22+
nur in einem kleinen Bereich, bei Eh > 0,9 und pH < 5, stabil ist, und die Speziation bei niedrigeren pH-Werten von drei- bis fünfwertigen Spezies dominiert wird.
Die Hydrolyse von Pu ist bei den hohen Oxidationsstufen (V und VI) stärker ausgeprägt als dies für die dreiwertigen Ln und An, wie Europium, Curium oder Americium der Fall ist. Im Allgemeinen ergibt sich für die Stärke der Hydrolyse- und Komplexbildung die Reihnfolge M4+ > MO2
2+ > M3+ > MO2
+. In Lösung bilden Pu(V) und Pu(VI) sogenannte Plutonyl-Ionen [O=Pu=O]+/2+. Diese sind linear angeordnet. Die Partialladung beträgt 2,2 bzw. 3,3.36
Abbildung 6: Die Pourbaix Diagramme ([Pu] = 10-4 M, [U] = 10-3 M, [NaCl] = 10-1 M, Eh-pH, in Luft) wurden mithilfe des geochemischen Codes Geochemist’s Workbench (Version 11.0.2, Modul Act2) erstellt.
Für die Modellierung von U und Pu wurde die aktuellste NEA thermodynamische Datenbank benutzt.
Pu(V) und Pu(IV) tendieren zur Disproportionierung, allerdings ist Pu(IV) deutlich stabiler als Pu(V), wobei die Kinetik aufgrund des Bildens und Brechens der Pu=O Bindung in Plutonyl-Ionen (PuO2
+ und PuO2
2+) relativ langsam ist. Dabei folgt die Disproportionierung von Pu(V) Gleichung (4.4):
(4.4)
Chemie der Actiniden
Daraus ergeben sich die Gleichungen (4.4a) und (4.4b), wobei (4.4a) aufgrund des Brechens der Pu=O Bindung relativ langsam abläuft, (4.4b) hingeben relativ schnell. Sobald beide Gleichungen (4.4a) und (4.4b) ihr Gleichgewicht erreicht haben, ist die Disproportionierung vollständig.37
(langsam) (4.4a) (schnell) (4.4b)
Die Disproportionierung von Pu(IV) folgt Gleichung (4.5),
(4.5)
wobei auch diese zweiteilige Reaktion in einer langsamen (4.5a) und einer schnellen (4.5b) Teilreaktion abläuft.
(langsam) (4.5a) (schnell) (4.5b)
Das radioaktive Nuklid 242Pu (Halbwertszeit: 3,75 × 105 Jahre), ein Alpha-Strahler, wurde in dieser Arbeit verwendet.
Uran besitzt die Elektronenkonfiguration [Rn]5f36d17s2 und seine Chemie ist, wie auch die des Plutoniums, im Vergleich zum Thorium deutlich komplexer. In wässriger Lösung können die Oxidationsstufen IV bis VI auftreten. Das chemische Verhalten unterscheidet sich dabei je nach Oxidationsstufe deutlich. U bildet wie Pu in den Oxidationsstufen V und VI ebenfalls Actinyl- Ionen, die sogenannten Uranyl-Ionen [O=U=O]+/2+. In den jeweiligen Oxidationsstufen verhalten sich die Elemente weitgehend analog.
Die Reaktionen der Actinyle in Lösung sind oftmals schon durch deren Größe (der Radius von z. B. Uranyl ergibt sich aus U-O Distanz von ~ 1,7 Å + Ionenradius eines Sauerstoffions von
~ 1,4 Å = ~ 3,1 Å)38 kinetisch gehemmt, speziell wenn diese zusätzlich hydratisiert sind.
Uran besitzt in wässrigen Lösungen ein komplexes Redoxverhalten, was unter anderem an den sehr unterschiedlichen Redoxpotentialen der unterschiedlichen Redoxpaare liegt. (z.B.
UVI/UV = 0,088; UVI/UIV = 0,267; UIV/UIII = -0,553; für 298,15 K in V vs. SHE in Reinstwasser).23,39 Sollte es zu Änderungen des Oxidationszustandes von U aufgrund von Änderungen der
Lösungseigenschaften wie pH-/Eh-Wert kommen (siehe Abbildung 6), so läuft diese Redoxtransformation sehr schnell ab, wenn es zu keinem Brechen der Uranylbindung kommt.
Kommt es zu diesem, läuft die Reaktion sehr langsam ab. Das heißt die Reaktionen (4.6a) und (4.6b) laufen schnell ab:
(4.6a)
(4.6b)
Während z. B. Reaktion (4.7) langsam abläuft:
(4.7)
Grund dafür ist das Brechen/Bilden der U-O-Bindungen des Uranyls, welche eine kinetische Barriere für die Redoxreaktion darstellen. Entsprechend der genannten großen Unterschiede der Redoxpotentiale verbleibt U(VI) unter oxidierenden Bedingungen in seinem prädominanten Oxidationszustand U(VI). Wie Abbildung 6 zeigt, dominiert UO22+
die Speziation bei Eh-Werten
> 0,2 unter sauren Bedingungen bis pH 4,2. Mit steigendem pH-Wert und unverändertem Eh-Wert dominieren verschiedene Hydroxo- sowie Ternäre Komplexe. Bei Eh-Werten < 0,2 und sehr sauren Bedingungen (kleiner pH 2,2) dominiert UCl3+, falls Cl- vorhanden ist, bzw. U(OH)2
2+. Bei pH- Werten > 2,2 und gleichzeitig kontinuierlich fallendem Eh-Wert dominiert eine amorphe UO2- Phase. Im Vergleich zu PuO2
2+, was ein kleines Stabilitätsfeld (Eh 0,9 - 1,1 V) in wässriger Lösung hat, weist UO2
2+ (Eh 0,2 - 1,1 V) also ein deutlich größeres Stabilitätsfeld auf. UO2
+ in wässrigen Lösungen, neigt zur Disproportionierung nach demselben Schema wie für PuO2
+ dargestellt wurde (Gleichung 4.4).
Das radioaktive Nuklid 238U (Halbwertszeit: 4,47 × 109 Jahre), ein Alpha-Strahler, wurde in dieser Arbeit verwendet.
4.2.3 Nanopartikel
Eine spezielle Eigenschaft der vierwertigen Actiniden Th, U, Np und Pu ist ihre Fähigkeit durch Olations- und/oder Oxolationsreaktionen (Gleichung 4.8a-b) Nanopartikel auszubilden, die auch als Eigenkolloide bezeichnet werden.40
Chemie der Actiniden
(4.8)
(4.8a - Olation) (4.8b - Oxolation)
Das Auftreten von Partikeln im Bereich < 1 µm in natürlichem Grundwasser ist hinreichend bekannt41-44 und aufgrund ihrer Größe und geringen Sedimentationsgeschwindigkeit45 können diese über lange Zeiträume in Suspension bleiben. Die Rückhaltung von Radionukliden/Kontaminanten wird durch Kolloide, suspendierte Partikel im nm- bis µm-Bereich, in der Regel negativ beeinflusst.46,47 Schwer- oder unlösliche Kontaminanten können an Kolloiden anhaften40,46,48 und somit über weite Strecken transportiert werden. Dabei spielt es keine Rolle ob es sich um anorganische oder organische Kolloide handelt.49,50 Für diesen Prozess müssen die Kontaminanten nicht selbst in der Lage sein, Eigenkolloide zu bilden. Wenn sich aus mobilen gelösten Ionen, zum Beispiel durch oberflächenvermittelte Prozesse,51 Nanopartikel bilden, die an Mineralen adsorbiert sind, wirkt dieser Vorgang zunächst retardierend. Sollten sich diese Nanopartikel allerdings von der Oberfläche lösen und in einen kolloidalen Zustand übergehen, so wird deren Mobilität erhöht.
Die Bildung von Nanopartikeln an Mineraloberflächen bedingt zuvor die Bildung von Dimeren und kleineren Oligomeren. Die Bildung der Dimere funktioniert in wässrigen Lösungen durch Kondensationsprozesse aus vorhandenen Hydrolysespezies. Dabei sind die Metallionen durch einen Hydroxidliganden (Gleichung 4.8a - Olation) oder einen Sauerstoffliganden (Gleichung 4.8b - Oxolation) verbrückt. Mit zunehmender Härte wird dabei die Oxolation favorisiert.31 Neben dem Hauptfaktor, der Härte, hängt der Kondensationspfad zusätzlich vom Hydrolyse-Komplex und den Lösungsbedingungen, wie Metallkonzentration und pH-Wert, ab.
4.3 Grenzflächenreaktionen von Actiniden auf Muskovit
4.3.1 Sorption von Th an der Basalfläche von Muskovit im Medium NaCl
In vorangegangenen Arbeiten von Schmidt et al.1 wurde das Sorptionsverhalten von Th im Kontakt mit der Basalfläche von Muskovit untersucht ([Th] = 0,1 mM, [NaCl] = 0,1 M, pH = 3,2).
Die Ergebnisse dieser Arbeit sollen für die Th-Experimente der vorliegenden Studie als Referenz dienen. Die Strukturanalyse wurde in situ mithilfe von SXS (siehe Kapitel 5.1.2) durchgeführt.
Zudem wurde die Oberflächenbelegung unabhängig mittels Alpha-Spektrometrie bestimmt. Die ermittelten Belegungen des oberflächenassoziierten Th betragen 0,15 ± 0,02 Th/AUC (Alpha- Spektrometrie) bzw. 0,40 ± 0,07 Th/AUC (RAXR). Die alpha-spektrometrisch ermittelten Oberflächenbelegungen in Abhängigkeit von der Th-Konzentration ([Thtot] = 1,0 × 10-6 – 5,0 × 10-3 M) können für niedrige Konzentrationen (bis 1 × 10-3 M) durch eine Langmuir-Verteilung darstellt werden, für höhere Konzentrationen ist die Belegung durch oberflächeninduzierte Oligomerisation deutlich erhöht.1
Abbildung 7: Messungen der Th(IV)-Aufnahme,1 wobei die Oberflächenbelegung durch Alpha-Spektrometrie ermittelt wurde und als Funktion der Th-Konzentation in Lösung dargestellt wurde ([Th]tot = 1,0 × 10-6 bis 4,9 × 10-3 M). Die best-fit Langmuir-Isotherme für die niedrigen Konzentrationen ist als durchgezogene rote Linie dargestellt. Die gestrichelte orange Linie zeigt den Trend der überschüssigen Th-Aufnahme durch oberflächeninduzierte Oligomerisation. Das obere Fenster zeigt die Daten bei niedrigen Konzentrationen von [Th]tot = 1,0 × 10-6 bis 1,0 × 10-3 M, welche für den Langmuir-Fit verwendet wurden.
Die Adsorptionsstruktur an der Fest-Flüssig-Grenzfläche (in situ) konnte als eine breit verteilte Spezies, mit einem Maximum im Abstand von 10,1 Å von der Mineraloberfläche, identifiziert werden. Dieser große Abstand zur Oberfläche kann nicht mit den klassischen Modellen der IS- und OS-Sorption erklärt werden und wird als Extended-OS-Komplex interpretiert.1,52,53 Das heißt, Th
Chemie der Actiniden
adsorbiert mit zwei intakten Hydrathüllen. In den meisten Fällen treten Extended-OS-Komplexe als minorer Anteil zusammen mit dominierenden IS- und OS-Komplexen auf (z.B. im Falle von zweiwertigen Kationen wie Sr2+ oder Pb2+),53 im Falle von Th-NaCl-Muskovit dominiert hingegen der Extended-OS-Komplex. Dies ist durch die hohe freie Hydratationsenthalpie des kleinen, hochgeladenen Th(IV)-Kations zu erklären (ΔGhyd (Th4+) = -5815 kJ/mol),54 welche signifikant höher ist als jegliche freie Hydratationsenthalpie von zweiwertigen Kationen, z.B. des am stärksten hydratisierten zweiwertigen Kations Be2+ (unter gleichen Bedingungen:
ΔGhyd (Be2+) = - 2395 kJ/mol)1,54 oder als die von dreiwertigen Kationen, z.B. Y3+
(ΔGhyd (Y3+) = - 3620 kJ/mol).55,56
4.3.2 Pu(IV)-Nanopartikel aus einer Pu(III)-Lösung.
Pu-Nanopartikel sind Inhalt von diversen aktuellen Forschungsstudien.40,46-48,51,57
Ausgangspunkt sind dabei häufig Standorte, an denen eine Kontamination durch Pu vorliegt und der Fokus auf Untersuchungen der Mobilität von Pu im Zusammenhang mit Nanopartikeln liegt.40,46,47 Andere Studien haben gezeigt, dass Aggregate von an sich schwerlöslichem PuIV zu einer erhöhten Löslichkeit führen können, entweder in Form von intrinsischen Kolloiden oder durch Adsorption an gelösten mineralischen Nanopartikeln.58-60
Die vorangegangene Studie von Schmidt et al.51 untersuchte die Bildung von Pu(IV)- Nanopartikeln auf der basalen (001) Fläche von Muskovit, ausgehend von einer Pu(III)-Lösung.
Alpha-Spektrometrie sowie RAXR-Messungen ergaben sehr hohe Pu-Belegungen auf der Muskovitoberfläche von jeweils 9,0 ± 0,5 bzw. 9,9 ± 1,2 Pu/AUC. Die Stabilität der Pu(III)-Lösung wurde anhand von UV/Vis-Spektroskopie über einen Zeitraum von mehr als einer Woche bestätigt.
Weder eine nachweisbare Pu(IV)-Konzentration (Nachweisgrenze bei spektraler Interferenz für eine 1 mm Zelle: 160 µM)61 noch Polymerphasen konnten nachgewiesen werden.62 Da nach einer Kontaktzeit von 24 h Pu(IV)-Nanopartikel an der Muskovitoberfläche nachgewiesen werden konnten, spielt die Oberfläche offensichtlich eine entscheidende Rolle im Prozess der Nanopartikelbildung. Nach Schmidt et al.51 findet die initiale Sorption vermutlich in Form einer Extended-OS-Sorption, ähnlich der im Th-NaCl System beobachteten, statt, bis die Konzentration einen notwendigen Schwellenwert überschreitet und über Hydrolyse- und Kondensationsprozesse die Bildung von Nanopartikeln beginnt. Entscheidend für den gesamten Prozess der Nanopartikelbildung sind die Anwesenheit des Minerals und das Gleichgewicht (GGW) zwischen Pu(III) und Pu(IV). Dabei wird dem GGW kontinuierlich Pu(IV) durch die Sorption/Nanopartikelbildung entnommen. Dadurch wird Pu(III) ständig zu Pu(IV) oxidiert, um den
GGW-Zustand beibehalten zu können. Der Prozess ist also durch die Oberfläche katalysiert. Die Studie lässt offen, wo die Oxidation von Pu(III) zu Pu(IV) stattfindet. Drei mögliche Mechanismen für die Bildung von Pu(IV)-Nanopartikeln sind vorgeschlagen: (A) Pu(III) wird in der Bulk-Lösung zu Pu(IV) oxidiert und anschließend adsorbiert Pu(IV) an der Mineraloberfläche. (B) Pu(III) adsorbiert an der Mineraloberfläche und wird an der Oberfläche zu Pu(IV) oxidiert. Beide Pfade (A) und (B) führen über Hydrolyse- und Kondensationsprozesse daraufhin zur Bildung von Oligomeren und letztendlich Nanopartikeln an der Oberfläche. Der dritte Pfad (C) basiert auf der Bildung der Nanopartikel aus im Lösungsgleichgewicht vorhandenem Pu(IV) direkt in Lösung, gefolgt von der Sorption dieser an der Mineraloberfläche. Pfad (C) ist auf Grundlage der bekannten Thermodynamik des Pu allerdings unwahrscheinlich.
4.3.3 Einfluss von Mineraloberflächen auf die Bildung von Pu(IV)
Die Bildung von Pu(IV) an Mineraloberflächen im Kontakt mit Lösungen die Pu in den Oxidationsstufen Pu(III) bis zu Pu(VI) beinhalten, wurde in vorangegangenen Arbeiten beobachtet.
Sehr viele Studien haben dabei die Relevanz von redox-aktiven Mineralen untersucht.63-72 Dass dieses Kriterium aber keineswegs eine Notwendigkeit für die Bildung von Pu(IV) darstellt, konnte wiederum in mehreren Arbeiten mit redox-inaktiven Mineralen gezeigt werden.51,73-76 An den Oberflächen der redox-aktiven Minerale ist häufig Eisen oder Mangan verfügbar, sodass ein direkter Elektronentransfer vom oder zum Mineral stattfindet. Wohingegen die redox-inaktiven Minerale zwar für die Reaktionen notwendig sind oder diese begünstigen, allerdings nicht direkt am Elektronentransfer teilnehmen können. Ähnlichkeiten mit der vorliegenden Arbeit liegen z.B. in der Studie über die Reduktion von Pu(V) zu Pu(IV) an Montmorillonit vor.75 Die Reduktion an der Grenzfläche des redox-inaktiven Montmorillonits verlief ebenfalls langsam und aufgrund der Abhängigkeit dieser Reaktionsgeschwindigkeit von pH und Ionenstärke, wurde vermutet, dass ein Protonen-vermittelnder Oberflächenprozesses involviert ist.
Die oberflächenvermittelte Bildung einer Phase auf einem Substrat kann durch epitaxiale Übereinstimmung erleichtert oder beeinflusst werden. Das heißt, die vorgegebene atomare Ordnung eines kristallinen Substrats wird auf die Phase, die sich an dessen Oberfläche bildet, übertragen. Ein solcher Effekt wurde für die Bildung von Pu-Nanopartikeln auf Goethit (FeOOH) beobachtet, wo Partikel mit Pu4O7-Struktur statt der üblichen PuO2-Struktur gefunden wurden.77

![Abbildung 6: Die Pourbaix Diagramme ([Pu] = 10 -4 M, [U] = 10 -3 M, [NaCl] = 10 -1 M, Eh-pH, in Luft) wurden mithilfe des geochemischen Codes Geochemist’s Workbench (Version 11.0.2, Modul Act2) erstellt](https://thumb-eu.123doks.com/thumbv2/1library_info/4565131.1599804/26.918.277.624.117.762/abbildung-pourbaix-diagramme-mithilfe-geochemischen-geochemist-workbench-erstellt.webp)
![Abbildung 7: Messungen der Th(IV)-Aufnahme, 1 wobei die Oberflächenbelegung durch Alpha-Spektrometrie ermittelt wurde und als Funktion der Th-Konzentation in Lösung dargestellt wurde ([Th] tot = 1,0 × 10 -6 bis 4,9 × 10 -3 M)](https://thumb-eu.123doks.com/thumbv2/1library_info/4565131.1599804/30.918.277.598.537.796/abbildung-messungen-aufnahme-oberflächenbelegung-spektrometrie-ermittelt-konzentation-dargestellt.webp)